Research group Prof. Dr. Nikos Doltsinis
Theory of Functional Nanostructures

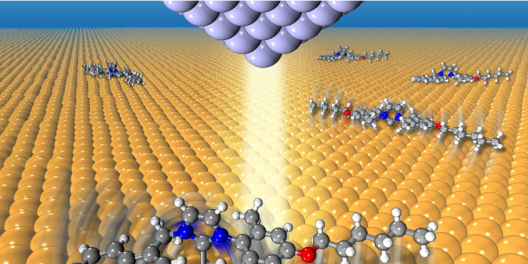
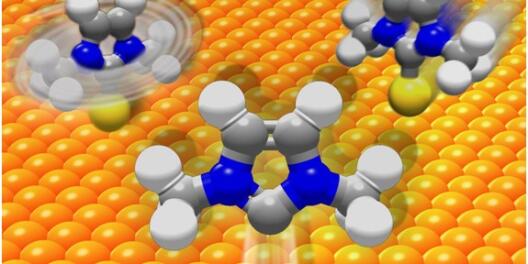
The research area of our group is computer simulations of the dynamics of molecules in the condensed phase, i.e. in liquids, clusters and solids. Different methods with different accuracy are used - from high-precision quantum mechanical methods to classical semiempirical methods. Their combination in the framework of multiscale models enables the simulation of physical processes over several orders of magnitude on the length and time scale.
We are active in the development of new simulation methods as well as in their application to current problems in physics and related fields. Topics for bachelor and master theses are currently available in the areas of "Acceleration methods for molecular dynamics simulations", "Light-driven materials", "Molecular electronics" and "Structural analysis of soft matter using atomic probe tomography".