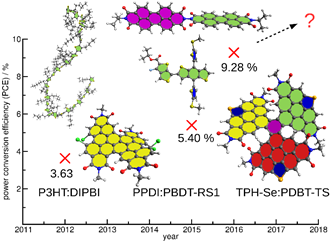
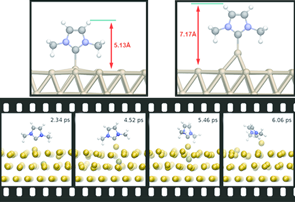
Research interests
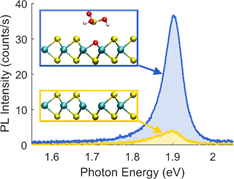
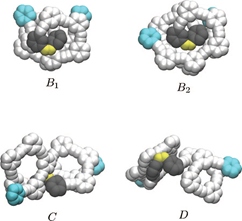
By approximating the Schrödinger equation in the context of density functional theory, the quantum mechanical properties of larger solids or molecules can be calculated accurately. Thus, in addition to chemical bonds, charge transfer processes and - by considering external fields - optical properties of real systems can be predicted.
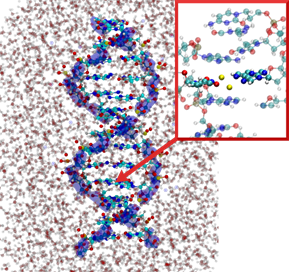
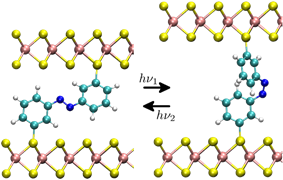
Beyond static properties, molecular dynamics simulations allow the study of dynamic processes. If the underlying potential is derived from electronic structure calculations, for example, the breaking of chemical bonds as well as light-induced isomerizations can be described. However, by using classical force fields, much larger systems, such as DNA strands in solution, can be simulated over several microseconds.