
Topics of research
Research Profile Prof. Dr. Bernhard Wünsch
At the Institute of Pharmaceutical and Medicinal Chemistry we are working on the development of novel ligands addressing receptors in the central nervous system and ion channels. In addition to pharmacologically active compounds, imaging probes, such as PET tracers and fluorescently labelled ligands, are designed, synthesized and used for molecular imaging of receptors and ion channels. Ligands covalently binding to target proteins are envisaged with the aim to analyze their binding pocket. Since the binding pockets of targets are composed of enantiomerically pure amino acids, the stereochemistry plays a central role in the projects. Physicochemical and pharmacokinetic properties, such as logD7.4 value, plasma protein binding and biotransformation are taken into account at a very early stage of the ligand development.
Key words:
Targets: NMDA receptor; CatSper; KCa3.1 channel; s receptors; KOR
Projects: Subtype selectivity; mechanism of action; imaging; fluorescence dyes; PET; in vitro ADME characterization; enantioselective synthesis; chemoenzymatic synthesis; structure activity relationships;
1. Development of subtype-selective NMDA receptor antagonists
The prototypical agonist N-methyl-D-aspartate gave name to the NMDA receptor. It is one of the ionotropic receptors for the excitatory neurotransmitter (S)-glutamate. Upon activation, the ligand gated ion channel allows the permeation of Na+- and Ca2+-ions into the neuron and the efflux of K+-ions. The NMDA receptor is characterized by multiple binding sites for ligands, which are able to modulate the open probability of the ion channel and thus the influx and efflux of ions. A typical NMDA receptor consists of two GluN1 and two GluN2 subunits. Four GluN2 subunits, which are termed GluN2A-D, are known, which determine the properties of the particular NMDA receptor subtype.
1.1. Ligands selectively addressing NMDA receptors with GluN2A subunit
In order to address NMDA receptors with GluN2A subunit, TCN-201 (Figure 1) is used as lead compound. TCN-201 binds in close proximity to the glycine binding site and shows high selectivity for NMDA receptors containing the GluN2A subunit. Four structural elements marked with boxes are extensively modified.
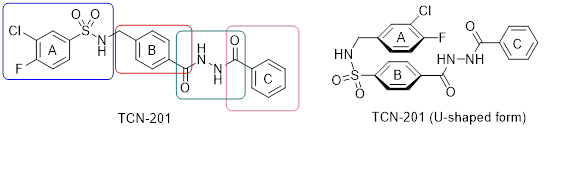
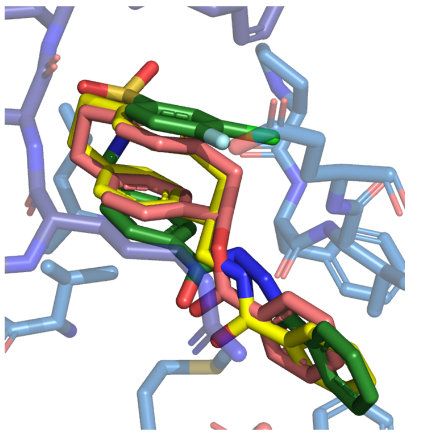
In the crystal structure, TCN-201 adopts an U-shaped conformation (Figure 1, right). In order to mimic this U-shaped conformation [2.2]paracyclophanes are designed, synthesized and pharmacologically evaluated. Figure 2 shows the very similar binding pose of TCN-201 and some [2.2]paracyclophanes.
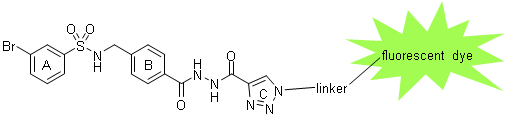
In order to image NMDA receptors with GluN2A subunit, ligands derived from TCN-201 connected with a fluorescent dye via a linker are designed, synthesized and biologically evaluated. Such fluorescent dye-labeled ligands are used to label NMDA receptors with GluN2A subunit in various assay systems, spheroids and brain slices. (Figure 3)
References
1. Systematic variation of the benzenesulfonamide part of the GluN2A selective NMDA receptor antagonist TCN-201. S. L. Müller, J. A. Schreiber, D. Schepmann, N. Strutz-Seebohm, G. Seebohm, B. Wünsch, Eur. J. Med. Chem. 2017, 129, 124 - 134. DOI 10.1016/j.ejmech.2017.02.018.
2. GluN2A-selective NMDA receptor antagonists: Mimicking the U-shaped bioactive conformation of TCN-201 by a [2.2]paracyclophane system. R. Steigerwald, T.-H. Chou, H. Furukawa, B. Wünsch, ChemMedChem 2022, e202200484. DOI: 10.1002/cmdc.202200484.
1.2. Ligands selectively addressing NMDA receptors with GluN2B subunit
Inhibition of NMDA receptors containing the GluN2B subunit can be used for the treatment of acute damage of neurons (e.g. stroke, epileptic seizures, injury) and chronic neurodegenerative disorders (e.g. Alzheimer’s Disease, Parkinson’s Disease, amyotrophic lateral sclerosis). NMDA receptors with GluN2B subunit present a special binding site within the amino-terminal domain, which is called ifenprodil binding site. Ligands interacting with the ifenprodil binding site can act as positive or negative allosteric modulators (PAMs or NAMs). Since this binding site is presented only by NMDA receptors with GluN2B subunit, NAMs at this binding site inhibit only this NMDA receptor subtype, resulting in an improved side effect profile.
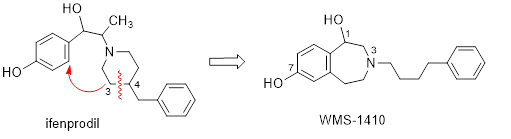
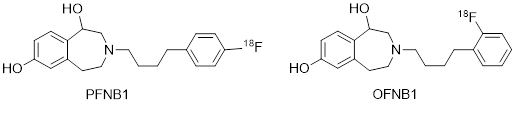
The class of tetrahydro-3-benzazepines is derived from formal reorganization of the prototypical NAM ifenprodil. (Figure 4) WMS-1410 shows almost the same affinity at NMDA receptors with GluN2B subunit as ifenprodil but considerably increased inhibitory activity. WMS-1410 reveals higher selectivity for GluN2B receptors over s and a receptors and increased metabolic stability. In the experimental autoimmune encephalomyelitis (EAE) mouse model of Multiple Sclerosis, WMS-1410 ameliorated the symptoms of the animals. An enantioselective synthesis of WMS-1410 and analogs leading to the (R)-configured eutomer is developed. Structural modifications of WMS-1410 combined with site directed mutagenesis and two-electrode voltage clamp experiments led to the development of the foot-in-the-door mechanism for inhibiting the NMDA receptor. The 18-F-labeled WMS-1410 analogs PFNB1 and OFNB1 represent very promising tracers for PET studies in vivo.
References
1. Enantiomerically pure 2-methyltetrahydro-3-benzazepin-1-ols selectively blocking GluN2B subunit containing N-methyl-D-aspartate receptors. B. Tewes, B. Frehland, D. Schepmann, D. Robaa, T. Uengwetwanit, F. Gaube, T. Winckler, W. Sippl, B. Wünsch, J. Med. Chem. 2015, 58, 6293 - 6305. DOI 10.1021/acs.jmedchem.5b00897.
2. Synthesis and pharmacological evaluation of enantiomerically pure GluN2B selective NMDA receptor antagonists. F. Börgel, M. Szermerski, J. A. Schreiber, L. Temme, N. Strutz-Seebohm, K. Lehmkuhl, D. Schepmann, S. M. Ametamey, G. Seebohm, T. J. Schmidt, B. Wünsch, ChemMedChem 2018, 13, 1580 - 1587. DOI: 10.1002/cmdc.201800214.
3. A common mechanism allows selective targeting of GluN2B subunit-containing N-methyl-D-aspartate receptors. J. A. Schreiber, D. Schepmann, B. Frehland, S. Thum, M. Datunashvili, T. Budde, M. Hollmann, N. Strutz-Seebohm, B. Wünsch, G. Seebohm, Commun. Biol. 2019, 2, 420 - 433. DOI: 10.1038/s42003-019-0645-6.
4. Identification and preclinical evaluation of a radiofluorinated benzazepine derivative for imaging the GluN2B subunit of the ionotropic NMDA receptor. A. Haider, I. Iten, H. Ahmed, A. Müller Herde, S. Gruber, S. D. Krämer, C. Keller, R. Schibli, B. Wünsch, L. Mu, S. M. Ametamey, J. Nucl. Med. 2019, 60, 259 - 266. DOI: 10.2967/jnumed.118.212134.
2. Ligands selectively inhibiting the Ca2+ channel CatSper on human sperm
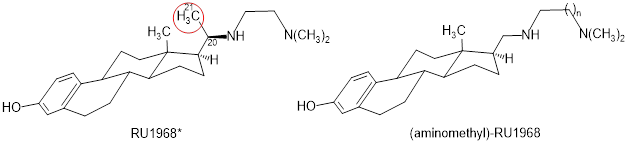
The sperm-specific Ca2+ channel CatSper (cation channel of sperm) controls the intracellular Ca2+ concentration and, thereby, the mobility and movement of sperm from many species. The steroidal ethylenediamine RU1968 represents a well-characterized, potent, and fairly selective cross-species inhibitor of CatSper. Due to its two additional centers of chirality in ring D and the attached side chain, RU1968 is difficult to synthesize, which hampers its broad use as a powerful tool for research. Simplified (aminomethyl)-RU1968 lacking the additional CH3 moiety in the side chain (red circle) is obtained diastereoselectively in enantiomerically pure form. Compared to RU1968, the homologous trimethylenediamine (n = 2) inhibited CatSper with two-fold enhanced potency. These aminomethyl analogs of RU1968 are enantiomerically pure, much easier to synthesize than RU1968 and exhibit higher CatSper inhibiting potency.
References
1. Synthesis and functional characterization of novel RU1968-derived CatSper inhibitors with reduced stereochemical complexity. T. Schierling, B. Tosi, C. Eisenhardt, S. Reining, C. Daniliuc, C. Brenker, T. Strünker, B. Wünsch, ACS Pharmacol. Transl. Sci. 2022, in press. DOI: 10.1021/acsptsci.2c00188.
3. Ligands selectively addressing the Ca2+ activated K+ channel KCa3.1
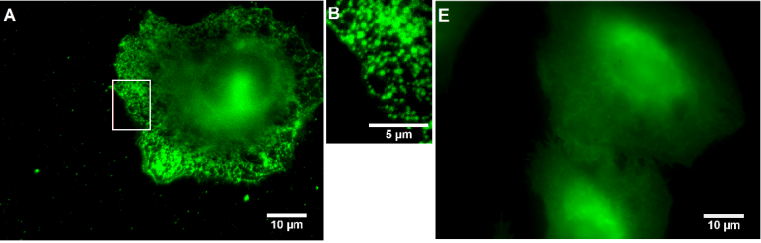
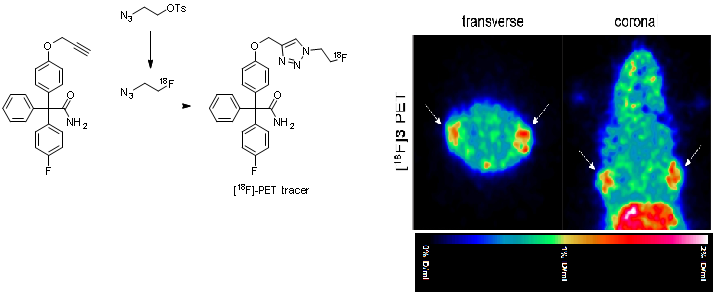
The Ca2+ activated K+ channel KCa3.1 is overexpressed in several human tumor cell lines, e.g. clear cell renal carcinoma, prostate cancer, non-small cell lung cancer. Highly aggressive cancer cells use this ion channel for key processes of the metastatic cascade such as migration, extravasation and invasion. Therefore, small molecules, which are able to image this KCa3.1 channel in vitro and in vivo represent valuable diagnostic and prognostic tool compounds. Senicapoc, which has been used in clinical trials for the treatment of sickle cell anemia, exhibits high inhibitory activity at the KCa3.1 channel. Incubation of tumor cells A549-3R with the senicapoc BODIPY conjugate led to the characteristic punctate staining pattern resulting from labeling of single KCa3.1 channels. The punctate staining was completely removed by preincubation with an excess of senicapoc confirming the specific interaction with KCa3.1 ion channel. (Figure 7)
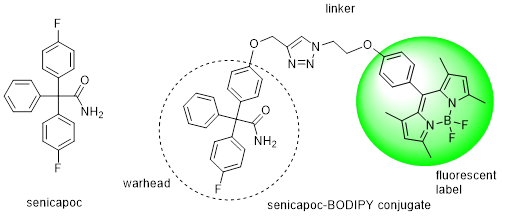
PET studies with [18F]fluoroethyltriazolyl substituted senicapoc ([18F]-PET tracer) led to imaging of KCa3.1 channels in lung adenocarcinoma cells in mice. The PET tracer was obtained by a,1,3-dipolar azide-alkyne cycloaddition (Click chemistry). (Figure 8)
References
1. Synthesis of small-molecule fluorescent probes for the in vitro imaging of calcium-activated potassium channel KCa3.1. K. Brömmel, S. Maskri, I. Maisuls, C. P. Konken, M. Rieke, Z. Pethö, C. A. Strassert, O. Koch, A. Schwab, B. Wünsch, Angew. Chem. Int. Ed. 2020, 59, 8277 - 8284 and Angew. Chem. 2020, 132, 8354 - 8361. DOI: 10.1002/anie.202001201 and 10.1002/ange.202001201.
2. Imaging of the calcium activated potassium channel 3.1 (KCa3.1) in vivo using a senicapoc-derived PET tracer. C. P. Konken, K. Heßling, I. Thale, S. Schelhaas, J. Dabel, S. Maskri, E. Bulk, T. Budde, O. Koch, A. Schwab, M. Schäfers, B. Wünsch, Arch. Pharm. 2022, e2200388. DOI: 10.1002/ardp.202200388.
4. Development of novel ligands for sigma receptors
The class of s receptors is subdivided into s1 and s2 receptors. s1 Receptors are involved in the modulation of various neurotransmitter systems and ion channels. A high density of s2 receptors has been detected in several human tumor cell lines. Therefore, ligands interacting with s1 receptors have a potential for the treatment of psychosis, depressions, epilepsy, neuropathic pain, cognition impairments, cocaine abuse and tumors. s2 receptor ligands are of increasing interest for the therapy and diagnosis of tumors. Our projects are devoted to the development of ligands binding with high affinity at s1 or s2 receptors and showing high selectivity against the other subtype and related receptors. For this purpose conformationally constrained ligands with defined stereochemistry are envisaged. Some potent s receptor ligands based on a bicyclic or spirocyclic scaffold or with a propellane skeleton are summarized in Figure 9.
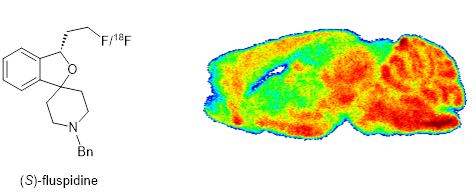
The most advanced s1 ligand of our group is (S)-fluspidine. The spirocyclic compound is analgesically active in the mouse capsaicin assay of neuropathic pain. The 18-F-labeled ligand can label regions in the central nervous system with high density of s1 receptors. In a small clinical study with patients suffering from major depressive disorder, the 18 F-labeled PET tracer [18F](S)-fluspidine showed an initial upregulation of the s1 receptor, which was downregulated in later phases of the disease. An enantioselective synthesis of (S)-fluspidine has been developed.
References
1. Synthesis of bicyclic s receptor ligands with cytotoxic activity. C. Geiger, C. Zelenka, M. Weigl, R. Fröhlich, B. Wibbeling, K. Lehmkuhl, D. Schepmann, R. Grünert, P. J. Bednarski, B. Wünsch, J. Med. Chem. 2007, 50, 6144 - 6153. DOI 10.1021/jm070620b.
2. Thiophene bioisosteres of spirocyclic s receptor ligands: Relationships between substitution pattern and s receptor affinity. C. Oberdorf, D. Schepmann, J. M. Vela, H. Buschmann, J. Holenz, B. Wünsch, J. Med. Chem. 2012, 55, 5350 - 5360. DOI 10.1021/jm300302p.
3. Synthesis, receptor affinity, and antiallodynic activity of spirocyclic sigma receptor ligands with exocyclic amino moiety. M. Bergkemper, E. Kronenberg, S. Thum, F. Börgel, C. Daniliuc, D. Schepmann, F. R. Nieto, P. Brust, R. F. Reinoso, I. Alvarez, B. Wünsch, J. Med. Chem. 2018, 61, 9666 - 9690. DOI: 10.1021/acs.jmedchem.8b01183.
4. Synthesis, pharmacological activity and structure affinity relationships of spirocyclic s1 receptor ligands with a (2-fluoroethyl) residue in 3-position. E. Große Maestrup, C. Wiese, D. Schepmann, P. Brust, B. Wünsch, Bioorg. Med. Chem. 2011, 19, 393 - 405. DOI 10.1016/j.bmc.2010.11.013.
5. Distinctive In vivo kinetics of the new sigma1 receptor ligands (R)-(+)- and (S)-(-)-18F-fluspidine in porcine brain. P. Brust, W. Deuther-Conrad, G. Becker, M Patt, C. K. Donat, S. Stittsworth, S. Fischer, A. Hiller, B. Wenzel, S. Dukic-Stefanovic, S. Hesse, J. Steinbach, B. Wünsch S. Z. Lever, O. Sabri, J. Nucl. Med. 2014, 55(10), 1730 - 1736. DOI 10.2967/jnumed.114.137562.
6. Short and atom-economic enantioselective synthesis of the s1-receptor ligands (S)- and (R)-fluspidine – important tools for positron emission tomography studies. P. Bunse, C. Schlepphorst, F. Glorius, M. Kitamura, B. Wünsch, J. Org. Chem. 2019, 84, 13744 - 13754. DOI: 10.1021/acs.joc.9b01882.
5. Development of novel KOR agonists
k Opioid receptors (KOR) belong to the class of opioid receptors. The clinically used analgesics are more or less selective µ receptor (MOR) agonists with the typical side effects including euphoria, respiratory depression, constipation, tolerance and dependence. In contrast to MOR agonists the side effect profile of KOR agonists is quite different. In particular the dangerous side effects such as respiratory depression and dependence are not associated with KOR analgesics. Moreover, KOR analgesics, which cannot pass the blood brain barrier, can be used for the treatment of inflammatory and painful situations in the periphery as well as for the treatment of pruritus. We are working on the development of conformationally constrained KOR agonists with high KOR affinity and high selectivity over MOR and DOR.
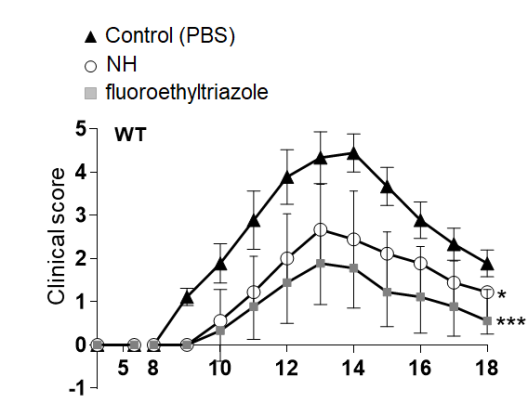
Recently, we developed perhydroquinoxaline-based KOR agonists shown in Figure 11. In a clinical phase I/IIa trial with patients with atopic dermatitis, dermal application of the methyl carbamate led to reduction of inflammation and itching. In the experimental autoimmune encephalomyelitis (EAE) mouse model of Multiple Sclerosis, the fluoroethyltriazole ameliorated the symptoms of the mice. (Figure 11 right).
References
1. Synthesis and pharmacological evaluation of 5-pyrrolidinylquinoxalines as a novel class of peripherally restricted k-opioid receptor agonists. C. Bourgeois, E. Werfel, F. Galla, K. Lehmkuhl, H. Torres-Gómez, D. Schepmann, B. Kögel, T. Christoph, W. Straßburger, W. Englberger, M. Soeberdt, S. Hüwel, H.-J. Galla, B. Wünsch, J. Med. Chem. 2014, 57, 6845 - 6860. DOI 10.1021/jm500940q.
2. Design and synthesis of enantiomerically pure decahydroquinoxalines as potent and selective k-opioid receptor agonists with anti-inflammatory activity in vivo. M. Soeberdt, P. Molenveld, R. P. M. Storken, R. Bouzanne des Mazery, G. J. Sterk, R. Autar, M. G. Bolster, C. Wagner, S. N. H. Aerts, F. R. van Holst, A. Wegert, G. Tangherlini, B. Frehland, D. Schepmann, D. Metze, T. Lotts, U. Knie, K.-Y. Lin, T.-Y. Huang, C.-C. Lai, S. Ständer, B. Wünsch, C. Abels, J. Med. Chem. 2017, 60, 2526 - 2551. DOI: 10.1021/acs.jmedchem.6b01868.
3. Development of novel quinoxaline-based k-opioid receptor agonists for the treatment of neuroinflammation. G. Tangherlini, D. V. Kalinin, D. Schepmann, T. Che, N. Mykicki, S. Ständer, K. Loser, B. Wünsch, J. Med. Chem. 2019, 62, 893 - 907. DOI: 10.1021/acs.jmedchem.8b01609.
6. Physicochemical and pharmacokinetic characterization of promising receptor ligands
Since physicochemical and pharmacokinetic properties of promising ligands should be taken into account at a very early stage during drug development, we have established methods to determine some ADME parameters in vitro. The logD7.4 value is determined by a micro shake flask method, which requires only very small amounts of compounds. The plasma protein binding is determined by a high performance affinity chromatography using a stationary phase coated with human serum albumin. Incubation of ligands with mouse or rat liver microsomes and NDDPH and/or additional cofactors such as UDP-GA gives insight into the metabolic stability of a candidate. Structures of metabolites are determined by mass spectrometry.
References
1. Pharmacokinetic properties of enantiomerically pure GluN2B selective NMDA receptor antagonists with 3-benzazepine scaffold. F. Börgel, F. Galla, K. Lehmkuhl, D. Schepmann, S. M. Ametamey, B. Wünsch, J. Pharm. Biomed. Anal. A 2019, 172, 214 - 222. DOI: 10.1016/j.jpba.2019.04.032.