
Ab-Initio Simulations
Despite the maturity of state-of-the-art commercial phase-change devices, the mechanism of electronic transport in the amorphous phase of phase-change materials and its properties are still not fully understood, especially at the nano-scale. Furthermore, the emergence of novel monatomic phase-change materials raises doubts about the validity of common transport models for the amorphous phase of phase-change materials.
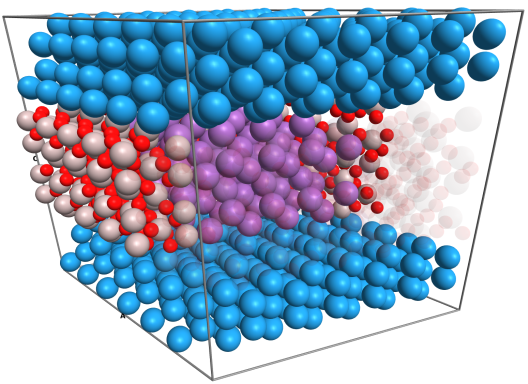
To this end, we complement our experimental work with ab-initio simulations based on density functional theory, which offer access to quantities that are not or not easily accessible in experiments. The amorphous phase is reached by rapidly quenching down in temperature from the liquid phase using ab-initio molecular dynamics simulations. This way, crystallization is avoided and the system is left in a meta-stable amorphous state. We study the electrical transport in this state on an atomistic level using the non-equilibrium Green's function method in combination with density functional theory.
We are particularly interested in scaling and interface effects, as future miniaturisation of phase-change devices may not only enable neuromorphic hardware capable of processing large amounts of information, but lead to increased device speeds, endurance, and decreased power consumption per operation.
For more information, please contact:
