Research interests - Prof. Dr. Michael Rohlfing
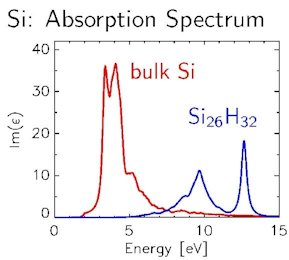
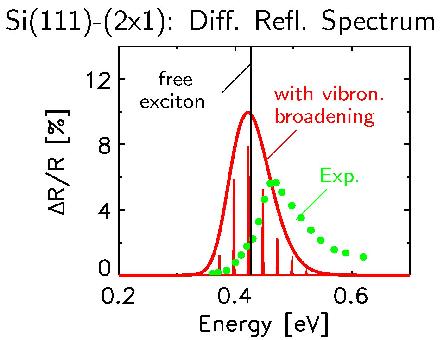
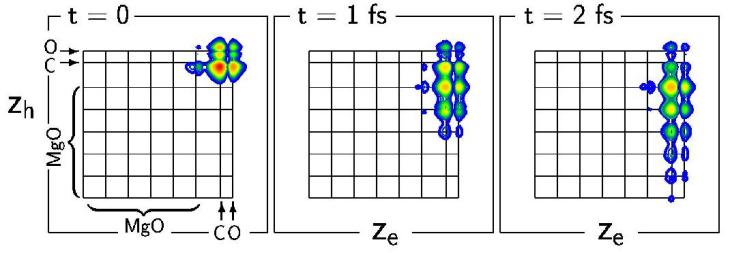
At the center of our work are excited electronic states in condensed matter. These states and their spectra play a central role in the understanding of optical properties, in the structural characterization of materials, and much more. We are particularly interested in systems characterized by quantum mechanical states on the atomic bond length scale. The properties of such nanoscopic materials are qualitatively far beyond the extended solid and can not be described by its characteristics. On the contrary, they require a microscopic theory which, as the smallest unit, attaches itself to the individual atom and its orbitals and is formulated as an ab initio theory, that is to say without specification of parameters. In addition, electronic states and their spectra are significantly influenced by many-particle effects (in particular electronic correlation), the careful handling of which by means of many-particle perturbation theory is a major aspect of this field of research.
An essential part of our methods is the symbiosis of fundamental physical concepts with numerical methods, i.e. the conversion of electronic structure theory into efficient computer algorithms. By means of such software we examine interesting aspects of different material classes. The methods can therefore be classified in the boundary between many-particle physics, numerical computer physics, and materials science.