
Electrolytes/Batteries
Polymer electrolytes (plus ionic liquids)
Mixtures of polymer electrolytes and ionic liquids
Solid polymer electrolytes have many advantageous properties such as very high energy density and convenient mechanical properties. This qualifies them as possible candidates for electrolytes in, e.g., lithium-ion batteries. Unfortunately, solid polymer electrolytes show poor ionic conductivity at room temperature. This problem has been overcome by adding room temperature ionic liquids ILs. Over the years molecular dynamics studies help to understand microscopic dynamics and provide many crucial insights about such systems.
We investigate ion transport mechanisms, their associations and its effect on ionic conductivity via molecular dynamics simulations. So far our analysis is focused on PEO/Li-TFSI/MPPY-TFSI systems. With state of the art force fields experimentally known structures and dynamical quantities (e.g. diffusion coefficients) can be reproduced satisfactorily.
Some key results: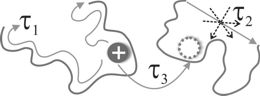
© ah 1. Since a Li-cation is coordinated with the polymer host, its transport properties are intimately connected with the polymer dynamics. We have identified three key Li transport mechanisms associated with distinct time scales: (i) intrachain motion along a chain (time scale τ1); (ii) polymer-segment motion translating the cation together with the polymer chain (time scale τ2, basically corresponding to the Rouse time); (iii) interchain hopping from one chain to another (time scale τ3). The resulting transport model can be solved analytically, reproducing simulation as well as experimental results.
© ah 2. ILs enhance the Li-diffusion by increasing the segment mobility of the host chain, that is, τ2 becomes shorter with higher IL-concentration. The key contribution of ILs is to prevent polymer crystallisation that results in high ionic conductivity at room temperature.
Ionic liquids
Mixture of ionic liquids and lithium salts
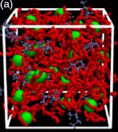
© ah Ionic liquids are a new class of materials. They have very promising properties for many applications, but only a few ionic liquids are well characterized until now. Despite the macroscopic characterization like diffusion coefficients, the microscopic properties are still not well unterstood. The advantages of this class include properties like low melting points, no significant vapor pressure and high dissolution power because of their electric charge.
In a collaboration with Stefano Passerini (KIT) and Oleg Borodin (U. S. Army Research Laboratory) we investigate via experimental and simulation techniques the transport properties of ionic liquids doped with lithium salts. As ionic liquids we use two different cations N-ethyl-N-methyl-imidazolium (emim) and lithium ions and two kinds of anions, namely bis(fluoro-sulfonyl)-imide (FSI) and bis(trifluoromethane-sulfonyl)-imide (TFSI). We are interested in different compositions of the anions and the influence on transport properties.
In collaboration with Dmitry Bedrov (University of Utah) we investigate the influence of different cations on lithium ion coordination and transport properties. For our analysis we choose one cation based on emim and one based on pyrrolidinium (N-methyl-N-propylimidazolium (pyr13)).Properties close to electrode interfaces
We investigate the molecular transport and reaction processes at the interface between electrode and electrolyte in lithium ion batteries using a combination of molecular dynamics (MD) and Monte-Carlo (MC) simulations. More specifically, we are interested in the microscopic mechanisms which lead to the initial formation of the Solid Electrolyte Interphase (SEI) and the formation of dendrites in lithium ion batteries.
Two complementary methodical approaches are pursued within our group. On the one hand, we are developing a lattice MC battery model that seeks to investigate the impact of molecular-scale parameters, such as charge transport properties, which have been derived from MD simulations, on macroscopic cell characteristics, for example quantified by the output voltage or current.
On the other hand, we are working on incorporating chemical reactivity into classical MD simulations by introducing reactive steps additionally to the MD steps. By doing so, we aim at developing a simulation method that incorporates chemical reactivity in a simple but realistic manner while retaining the time and length scales of standard MD simulations.Disordered materials
Supercooled liquids in (metastable) equilibrium / PEL approach
© ah For many years we have studied the dynamics of supercooled liquids. One way of charactering the dynamics is via mapping on the properties of the underlying potential energy landscape (PEL). In this framework the complex dynamics corresponds to jumps between different minima on the PEL. It turns out that the jump rates are strongly related to the potential energy of the corresponding minimum. Furthermore, by introducing some coarse graining procedure one can define sets of minima (metabasins) such that the dynamics between metabasins can be described as a simple random-walk. In this framework many important questions can be answered. What is the reason for the dramatic slowing down of glass-forming liquids? Why is silica a strong glass-forming system, i.e. shows simple Arrhenius-behavior? What is the reason why the viscosity and the diffusivity are decoupled for many glass-forming systems? What determines the emergence of slow and fast regions, i.e. the presence of dynamic heterogeneities? How do adjacent regions in the supercooled liquid communicate with each other? Within this approach we aim to obtain a far-reaching understanding of the very nature of the glass transition.
Rheology of supercooled systems and glasses
Applying a shear strain to a supercooled liquid or a glassy system shows interesting non-linear properties: Changes of the viscosity (e.g. shear thinning) as well as aging or rejuvenation. For this purpose we implement Lees-Edwards boundary conditions.
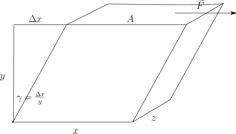
© ah Our focus lies in the description of these phenomena within the Potential Energy Landscape (PEL) framework. We therefore perform molecular dynamics simulations on small systems with our own simulation software written in C++, which enables us to use a special minimization algorithm to quickly gather information about the inherent structures (IS) the system visits. From this data we extract statistical properties and try to make a statistical model describing the observed macro-rheological behaviour. Of key interest in this description is the role of metabasins. To describe macroscopic systems, we also investigate the influence of finite size effects and the coupling of small systems to form a larger system.
Membranes/Biomolecules
Lipid membranes

© ah Rafts that are formed as a result of cooperation of saturated lipids with cholesterol or of saturated lipids with proteins or peptides are suggested to participate in various functions of the cell. The methods of MD simulations can provide important information on the driving forces of raft formation and properties of lipid phases. Comparison of the MARTINI coarse-grained (CG) bilayer system with an equivalent atomistic system (UA) comprised of DPPC, unsaturated dilinoleyl phosphatidylcholine (DUPC) lipids and cholesterol (CHOL) shows a very similar behavior concerning the nature of the phase separation.
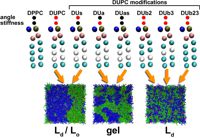
© ah We systematically vary the properties of CG lipids and CHOL to study the molecular requirements for the raft formation. It turns out that the unmixing is largely driven by van der Waals interactions between DPPC - CHOL/DPPC pairs and is nearly independent of the entropy of the DUPC chains. Furthermore by substituting the CHOL with shorter and stiffer DPPC-like molecules one observes unmixing similar to DPPC/DUPC/CHOL system which suggests the rigid and flat structure of CHOL to play a key role in raft formation.
We also put major efforts into the mapping of microscopically described mixtures of lipids and proteins onto 2D lattice models. A key question deals with the minimum number of the degrees of freedom to semi-quantitatively reproduce the microscopic system. In this way we aim to simulate complex mixtures of lipids and proteins in the near future.Interaction of proteins and lipid membranes
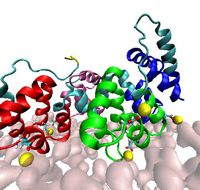
© ah With help of the HPC resources of the University of Muenster (PALMA) we actively investigate the properties of binding of certain surface proteins to a bilayer surface mediated by calcium ions where such protein-membrane interactions may induce raft formations. Jointly with experimental groups we investigate the interaction of lipids with proteins of the Annexin family.
Protein-protein interaction
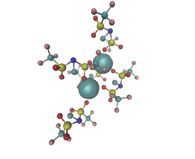
In collaboration with the group of Prof Mootz we investigate properties of the SIM/SUMO protein ligand complexes. The associaton of different SIM peptide chains and the SUMO protein oligomers plays a central role in posttranslational modification of proteins. We employ atomistic molecular dynamics simulations to understand the structure and dynamics of these complexes. Furthermore we use enhanced sampling methods to calculate the binding free energy of the these complexes.
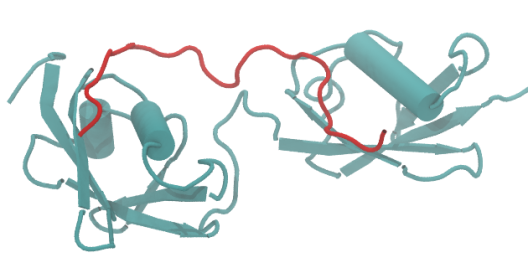
© ak The image shows a diSUMO (turquoise) protein in complex with a two SIM containing peptide chain (red). For example we want to find out what impact the complexation has on the dynamics of the SUMO subunits of the diSUMO relative to each other and what favorite conformation the SIM peptide chain adopts in the complex.
Structure formation on surfaces
Lipophilic molecules
Structure formation of lipophilic molecules on surfaces
Thin film deposition process is evolving continuously due to its huge technological importance in the field of organic field effect transistors (OFET), organic thin film transistors (OTFT), light emitting diodes (LED) etc. where different organic materials with conjugated π- orbitals are used as adsorbates. The efficiency of these nanoscale molecular devices depends on the molecular structure. In this project we investigate the influence of deposition rate and substrate temperature on the structure formation of substituted adenine nucleobases (lipophilic molecules). Experimentally, the emergence of two phases is observed (interdigitated and lamellar phase).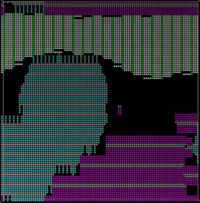
© ah Via appropriately devised lattice models this behavior can be reproduced and the non-equilibrium principles behind these observations can be clarified.
2D silica systems
Understanding the properties of two-dimensional silica systems
A new honeycomb like stable allotrope of silica has been recently prepared experimentally on Ru and graphene support. These experimental results have major impact in the field of semiconductors but serve as a very interesting model system to study supercooled liquids and glasses. It is a bi-layer structure with a hexagonal framework of Silicon-Oxygen bonds in each layer. Depending on preparation condition and cooling rates it is possible to generate crystalline and amorphous structures and sometimes both in the same layer; making it the world's thinnest glass former. A mysterious symmetry between upper and lower layer is present in both the crystal and amorphous form. Due to its two-dimensional nature, it is possible to map the position of the atoms in real space with STM or SPM methods. The ring statistics in amorphous phase has remarkable similarity with Zacharisen's prediction of glass structure of silica.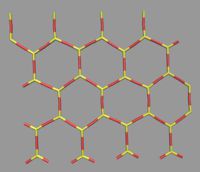
© ah We have developed a 2D silica type force-field that has the ability to generate the structural properties of two-dimensional silica. With the aid of molecular dynamics simulation, we are studying the structural and dynamic properties of this model at different conditions. We also plan to study how the various properties of bilayer silica depend, e.g., on the addition of metal ions.
Formation of designed patterns
Particles on close-by stripes
Concepts in simulation methodology and free energy calculations
Markov state modelling
Wang-Landau optimization
Soft matter assembly
Micelle formation
Frame-guided assembly of micelles and vesicles
The formation of self assembled structures such as micelles has been intensively study and is well understood. Recent studies use a new approach of vesicle formation by starting with a molecular frame (based, e.g., on DNA) which serves as a basis for new micelles/vesicles. In this project we develop a simple but powerful model for amphiphilic molecules and study the thermodynamcis and kinetics of micelle formation via stochastic simulations. The insights we acquire will then be applied to coarse grained MARTINI simulations of real systems.
Agglomeration properties of Janus particlesSports statistics
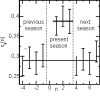
© ah