PALMA-NG
Content
Overview
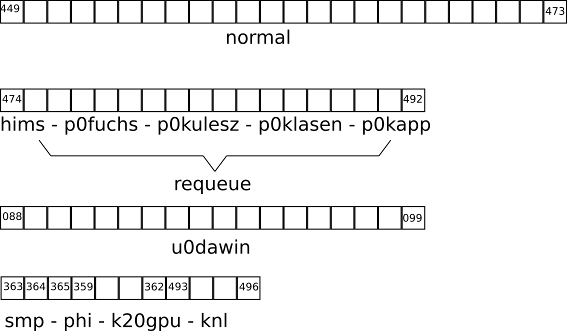
palma3 is the login node to a newer part of the PALMA system. It has various queues/partitions for different purposes:- k20gpu: Four nodes equipped with 3 K20 nVidia Tesla accelerators each
- normal: 29 nodes with 32 Broadwell CPU cores (64 threads) each and 128 GB RAM.
- knl: Four nodes with a Xeon Phi Knights Landing accelerator
- requeue: Job in this queue will run on the nodes of the above mentioned 18 nodes. If your jobs are running on one of the exclusive nodes while jobs are put in there, your job will be terminated and requeued, so use with care.
- express: A partition for short running (test) jobs with a maximum walltime of 2 hours.
- p0fuchs: 8 nodes for exclusive usage
- p0kulesz: 4 nodes for exclusive usage
- p0klasen: 1 nodes for exclusive usage
- p0kapp: 1 nodes for exclusive usage
- hims: 4 nodes for exclusive usage
Software/The module concept
The software on palma-ng can be accessed via modules. These are small script that set environment variables (like PATH and LD_LIBRARY_PATH) pointing to the locations where the software is installed (this is mostly on network drives so that the software is available on every node in the cluster). The module system we use here is LMOD| Command (Short- and Long-form) | Meaning |
|---|---|
| module av[ailable] | Lists all currently available modules |
| module spider | List all available modules with their description |
| module spider modulename | Show the description of a module and give a hint, which modules have to be loaded to make it available. |
| module li[st] | Lists all modules in the actual enviroment |
| module show modulname | Lists all changes caused by a module |
| module add modul1 modul2 ... | Adds module to the current environment |
| module rm modul1 modul2 ... | Deletes module from the current environment |
| module purge | Deletes all modules from current environment |
- foss/2016b and foss/2017a GCC with OpenMPI
- intel/2016b and intel/2017a Intel Compiler with Intel MPI
- goolfc/2016.10 Only on the k20gpu nodes for CUDA
module add intel/2016b module avand you will see the software that has been compiled with this version. Alternatively you can use the "module spider" command.
Using the module command in submit scripts
This is only valid for the u0dawin queue If you want to use the module command in submit scripts, the linesource /etc/profile.d/modules.sh; source /etc/profile.d/modules_local.shhas to be added before. Otherwise, just put the "module add" commands in your .bashrc (which can be found in your home-directory).
Monitoring
Ganglia
The batch system
The batch system on PALMA3 is SLURM, but there is a wrapper for PBS installed, so most of your skripts should still be able to work. If you want to switch to SLURM, this document might help you: https://slurm.schedmd.com/rosetta.pdf- The first line of the submit script has to be #!/bin/bash
- A queue is called partition in terms of SLURM. These terms will be used synonymous here.
- The variable $PBS_O_WORKDIR will not be set. Instead you will start in the directory in which the script resides.
- For using the "module add" command, you will have to source some scripts first: "source /etc/profile.d/modules.sh; source /etc/profile.d/modules_local.sh"
Submit a job
Create a file for example called submit.cmd#!/bin/bash # set the number of nodes #SBATCH --nodes=1 # set the number of CPU cores per node #SBATCH --ntasks-per-node 8 # set a partition #SBATCH --partition normal # set max wallclock time #SBATCH --time=24:00:00 # set name of job #SBATCH --job-name=test123 # mail alert at start, end and abortion of execution #SBATCH --mail-type=ALL # set an output file #SBATCH --output output.dat # send mail to this address #SBATCH --mail-user=your_account@uni-muenster.de # In the u0dawin queue, you will need the following line source /etc/profile.d/modules.sh; source /etc/profile.d/modules_local.sh # run the application ./programYou can send your submission to the batch system with the command "sbatch submit.cmd" A detailed description can be found here: http://slurm.schedmd.com/sbatch.html
Starting jobs with MPI-parallel codes
mpirun will get all necessary information from SLURM, if submitted appropriately. If you for example want to start 128 MPI ranks distributed to four nodes, you could do this the following way:srun -p normal --nodes=2 --ntasks=128 --ntasks-per-node=64 --pty bash mpirun ./programor for an non-interactive run or put those parameters in the batch script. For starting hybrid jobs (meaning that they are using MPI and OpenMP parallelization at the same time), you can use the --cpus-per-task switch.
srun -p normal --nodes=2 --ntasks=64 --ntasks-per-node=32 --cpus-per-task=2 --pty bash OMP_NUM_THREADS=2 mpirun ./program
Using GPU resources
The k20gpu queue features 4 nodes with 3 K20 nVidia Tesla accelerators each. To use one of these the following option must be present in your batch script:#SBATCH --gres=gpu:1It is also possible to use more than one. Additionally it is also possible to specify the type of GPU you want to work on. At the moment there are the following types:
- kepler: the standard type which can be used for most calculations
- kepler_benchmark: an alternative type if you want to use the second and third GPU without the first
#SBATCH --gres=gpu:kepler:1
Show information about the queues
scontrol show partition
Show information about the nodes
sinfo
Running interactive jobs with SLURM
Use for example the following command:srun --partition express --nodes 1 --ntasks-per-node=8 --pty bashThis starts a job in the u0dawin queue/partition on one node with eight cores.
Information on jobs
List all current jobs for a user:squeue -u <username>List all running jobs for a user:
squeue -u <username> -t RUNNINGList all pending jobs for a user:
squeue -u <username> -t PENDINGList all current jobs in the normal partition for a user:
squeue -u <username> -p normalList detailed information for a job (useful for troubleshooting):
scontrol show job -dd <jobid>Once your job has completed, you can get additional information that was not available during the run. This includes run time, memory used, etc.
To get statistics on completed jobs by jobID:
sacct -j <jobid> --format=JobID,JobName,MaxRSS,ElapsedTo view the same information for all jobs of a user:
sacct -u <username> --format=JobID,JobName,MaxRSS,ElapsedShow priorities for waiting jobs:
sprio
Controlling jobs
To cancel one job:scancel <jobid>To cancel all the jobs for a user:
scancel -u <username>To cancel all the pending jobs for a user:
scancel -t PENDING -u <username>To cancel one or more jobs by name:
scancel --name myJobNameTo pause a particular job:
scontrol hold <jobid>To resume a particular job:
scontrol resume <jobid>To requeue (cancel and rerun) a particular job:
scontrol requeue <jobid>--


| I | Attachment | History | Action | Size | Date | Who | Comment |
|---|---|---|---|---|---|---|---|
| |
llview.png | r2 r1 | manage | 49.3 K | 2016-12-06 - 12:31 | HolgerAngenent | |
| |
palma-ng_batchsystem.png | r2 r1 | manage | 23.6 K | 2017-03-02 - 12:53 | HolgerAngenent |
Topic revision: r28 - 2018-04-17 - HolgerAngenent


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Ideas, requests, problems regarding ZIVwiki? Send feedback
Datenschutzerklärung Impressum