
ML in der theoretischen Analyse und Simulation molekularer Systeme
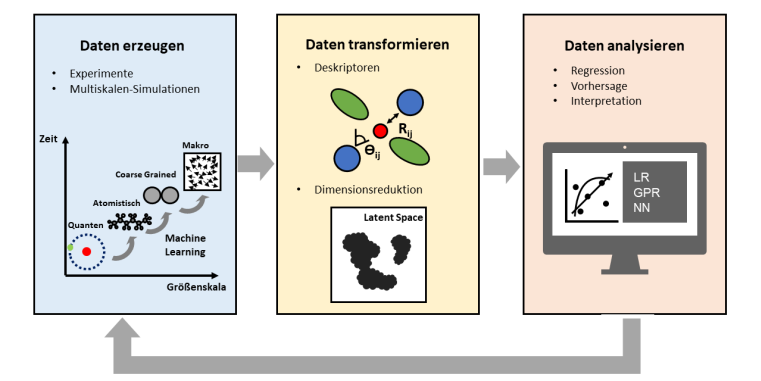
In den letzten Jahren hat der Einsatz von Methoden des Maschinellen Lernens (ML) im Bereich der (theoretischen) Analyse molekularer Systeme zunehmend an Bedeutung gewonnen. Dabei unterstützt ML sowohl die Datenauswertung von Experimenten als auch die Datenerzeugung mithilfe verschiedener Simulationsmethoden von der quanten-chemischen bis hin zur makroskopischen Skala. Im Bereich von Dichtefunktionaltheorie und Molekulardynamik-Simulationen übertreffen ML-Modelle teilweise schon jetzt klassische Methoden in den Punkten Genauigkeit und Simulationszeit um ein Vielfaches. Dabei ersetzen ML-Modelle die klassischen Methoden nicht immer, sondern erweitern diese und stellen neue Ansätze zur Verfügung.
Im Bereich der Datenauswertung ermöglichen Clusteralgorithmen und Regressionsmethoden das Erkennen hochkomplexer Zusammenhänge in hochdimensionalen Räumen. Bilderkennungssoftware kann die Auswertung von Spektren vereinfachen oder vollständig übernehmen. Letztendlich lassen sich so hochautomatisierte Prozesse erstellen, die nur ein minimales Eingreifen des Menschen erfordern und so Hochdurchsatz-Experimente ermöglichen, bei denen ein Computer mithilfe von Active Learning darüber entscheidet, welche Experimente als nächstes durchgeführt werden.
In den nächsten Jahren ist eine steigende Zahl von Anwendungen und eine weitere Erforschung der Möglichkeiten des Maschinellen Lernens im Bereich der Analyse molekularer Systeme erwarten.
Lehrveranstaltungen
In dem Modul „Maschinelles Lernen in der theoretischen Analyse und Simulation molekularer Systeme“ werden in einer jeweils im Wintersemester stattfindenden Vorlesungsreihe sowie einem zugehörigen Praktikum anhand ausgewählter Beispiele verschiedene Methoden des Maschinellen Lernens vermittelt. Die Vorlesung ist für Einsteiger auf dem Gebiet des Maschinellen Lernens geeignet, die grundlegendes Interesse an der Theorie mitbringen (Vorkenntnisse z.B. aus den Mastermodulen „Theoretische Chemie“, „Quantentheorie des Festkörpers“ oder „Elementare Anregungen in Festkörpern“ sind hilfreich, aber nicht zwingend notwendig).
Unterteilt ist die Vorlesung in mehrere Abschnitte, die sich jeweils mit aktuellen Methoden und Forschungsfragen in unterschiedlichen Teilbereichen beschäftigen. Ein Teil der Vorlesung beschäftigt sich mit der Frage, wie Maschinelles Lernen dazu beitragen kann, Austausch-Korrelations-Funktionale im Bereich der Dichtefunktionaltheorie zu verbessern. Ein anderer Teil beschäftigt sich mit Machine Learning Potentialen (MLP), welche genutzt werden können, um verschiedene quantenchemische und makroskopische Eigenschaften eines Moleküls vorherzusagen, die eine Alternative zu klassischen Kraftfeldern darstellen, und mit denen sich Coarse-Grained-Potentiale aus atomistischen Potentialen ableiten lassen.
Bei der Auswertung von Daten und Trajektorien können Clusteranalysen und Dimensionsreduktion neue Erkenntnisse über komplexe Zusammenhänge liefern. Anhand eines Beispiels zur Charakterisierung von Struktur-Dynamik-Beziehungen werden unterschiedliche Algorithmen vorgestellt und verglichen. Darüber hinaus wird Active Learning als eine Methode vorgestellt, um Systemeigenschaften zu optimieren und die Erzeugung neuer Daten zu automatisieren.
Die in der Vorlesung angesprochenen Themen werden im Rahmen eines begleitenden Praktikums vertieft, in dem die verschiedenen Methoden selbst ausprobiert werden können.
Die Vorlesung und das Praktikum finden in Präsenz statt. Die Kursinhalte (Folien, Skripte und Jupyter- Notebooks) können darüber hinaus jederzeit auch online studiert werden. Für den Erhalt von Leistungspunkten muss die Teilnahme an Vorlesung und Praktikum erfolgen.
Die Veranstaltung findet erstmals im Wintersemester 2023/24 statt.