|
|
|
Free Neuropathology 6:8 (2025) |
|
Original Paper |
|
Depletion of nuclear cytoophidia in Alzheimer’s disease |
|
Alyona Ivanova 1 , David G. Munoz 2 , John Woulfe 3,4 |
|
|
Corresponding author:
|
|
Additional resources and electronic supplementary material: supplementary table 1 |
|
Submitted: 28 January 2025
|
|
Keywords: Alzheimer’s disease, Cytoophidia, Phosphoribosyl pyrophosphate synthetase, Intranuclear rodlets, Purines |
|
Abstract There is considerable evidence for a role for metabolic dysregulation, including disordered purine nucleotide metabolism, in the pathogenesis of Alzheimer’s disease (AD). Purine nucleotide synthesis in the brain is regulated with high fidelity to co-ordinate supply with demand. The assembly of some purine biosynthetic enzymes into linear filamentous aggregates called “cytoophidia” (Gk. Cellular “snakes”) represents one post-translational mechanism to regulate enzyme activity. Cytoophidia comprised of the nucleotide biosynthetic enzymes inosine monophosphate dehydrogenase (IMPDH) and phosphoribosyl pyrophosphate synthetase (PRPS) have been described in neuronal nuclei (nuclear cytoophidia; NCs). In light of the involvement of purine nucleotide dysmetabolism in AD, the rationale for this study was to determine whether there are disease-specific qualitative or quantitative alterations in PRPS cytoophidia in the AD brain. Double fluorescence immunostaining for PRPS and the neuronal marker MAP2 was performed on tissue microarrays of cores of temporal cortex extracted from post-mortem tissue blocks from a large cohort of participants with neuropathologically confirmed AD, Lewy body disease (LBD), progressive supranuclear palsy, and corticobasal degeneration, as well as age-matched cognitively unimpaired control participants. The latter group included individuals with substantial beta-amyloid deposition. NCs were significantly reduced in frequency in AD samples relative to those from controls, including those with a high beta-amyloid load, or participants with LBD or 4 repeat tauopathies. Moreover, double staining for PRPS and hyperphosphorylated tau revealed evidence for an association between NCs and neurofibrillary tangles. The results of this study contribute to our understanding of metabolic contributions to AD pathogenesis and provide a novel avenue for future studies. Moreover, because PRPS filamentation is responsive to a variety of drugs and metabolites, they may have implications for the development of biologically rational therapies. |
|
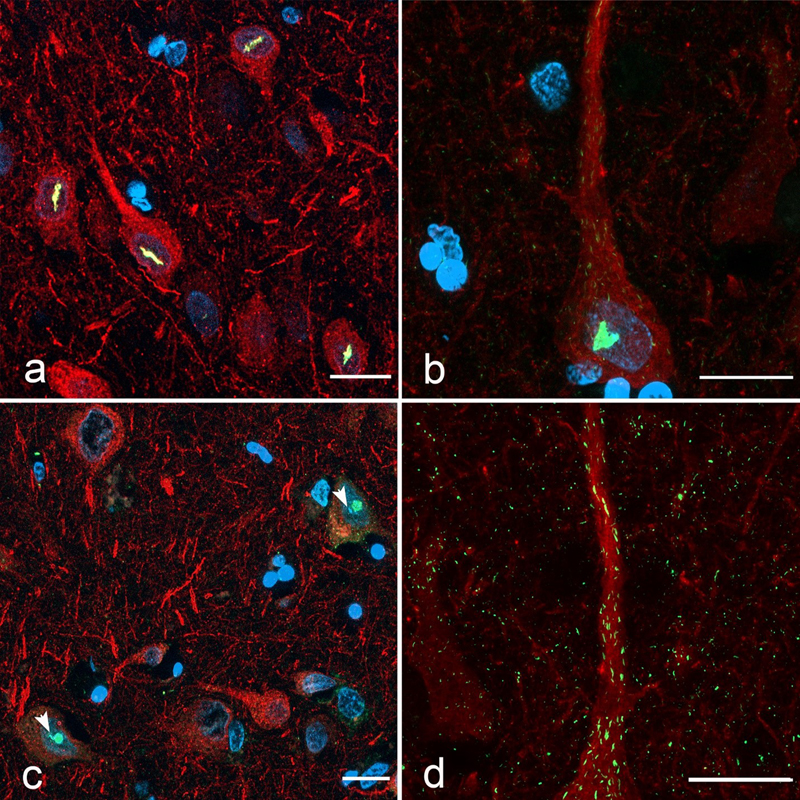
Introduction Alzheimer's disease (AD) is a devastating neurodegenerative disease characterized clinically by inexorable, progressive impairments in memory, judgement, and reasoning. The disease is characterized histopathologically by the buildup of beta-amyloid plaques and the presence of neurofibrillary tangles made up of phosphorylated tau protein. Although genetic evidence supports a critical role for beta-amyloid in the pathogenesis of familial forms of AD, the pathogenetic mechanisms underlying the far more common sporadic form of AD remain enigmatic. There is growing evidence for a role for metabolic dysregulation in AD, including nucleotide dysmetabolism, at early stages of AD independent of neurofibrillary tangles and β-amyloid plaques [1, 3, 26, 29–31]. There is deregulated expression of a number of purine metabolic enzymes in AD as well as significantly altered levels of metabolites including dGMP, glycine, xanthosine, inosine diphosphate, guanine, and deoxyguanosine [3]. The brain is critically dependent on an uninterrupted supply of purine nucleotides (AMP, ADP, ATP, GMP, GDP, GTP). They are assembled into RNA and DNA, provide energy for cells, are used as signalling molecules, and are incorporated into co-enzymes. Accordingly, inborn errors of purine nucleotide metabolism markedly, and relatively selectively impact the nervous system, resulting in severe neurodevelopmental diseases [16, 27, 38, 42, 51, 52, 66]. The synthesis of purine nucleotides is carefully regulated to calibrate supply with cellular demand. In response to purine limitation or a variety of manipulations, enzymes involved in the de novo purine pathway are assembled by liquid-liquid phase separation into microscopically visible, supramolecular membraneless organelles comprised of nine enzymes called "purinosomes" [54]. By virtue of the physical proximity of their constituent enzymes, these "metabolon" structures more efficiently catalyze the generation of AMP and GMP in response to metabolic demand [53]. In addition to purinosomes, a subset of nucleotide-synthesizing enzymes are subject to an additional level of supramolecular regulation; the formation of linear filamentous assemblies as a biological regulatory mechanism to co-ordinate enzyme activity with cellular requirements [5, 28, 41, 58]. The relationship between filament formation and enzyme activity is complex as both active and inactive multimeric conformers are present within individual filaments [28]. Current consensus supports a model whereby filament formation correlates with increased enzyme activity by resisting feedback inhibition by enzyme products [28]. This adds nuance to purinosome function; permitting ongoing nucleotide synthesis under conditions of increased nucleotide demand, even when purines are plentiful. Individual enzyme filaments can aggregate by side-to-side lateral alignment to form larger, mesoscale filament bundles, which have been variously referred to simply as "filaments", "rods and ring structures" or "cytoophidia" (Greek for "cellular snakes"; reviewed in [12, 40]). The formation of these structures at steady state and in response to metabolic perturbations has been described in a variety of cell types both in vitro and in vivo [12]. These filament bundles have been described both in the cytoplasm and the nucleus of a variety of cell types, including mammalian neurons [50]. We recently demonstrated the presence of cytoophidia immunoreactive for inosine monophosphate dehydrogenase (IMPDH) within the nuclei of neurons in the human brain [63, 64]. IMPDH is the rate-limiting enzyme in the de novo purine biosynthesis pathway. It is encoded by two genes, IMPDH1 and IMPDH2 [48]. These nuclear cytoophidia (NCs) are also immunoreactive for the nucleotide-synthesizing enzyme phosphoribosyl pyrophosphate synthetase (PRPS) [50, 63]. PRPS is a key enzyme in the de novo purine biosynthesis pathway as it catalyses one of the first steps; the production of phosphoribosyl pyrophosphate (PRPP). Humans contain three isoforms of PRPS (PRPS1, 2, and 3). In light of evidence for the involvement of nucleotide metabolism in AD pathogenesis cited above and the important role of cytoophidia formation in tuning metabolic enzymatic activity, we were interested in determining whether there are disease-specific alterations in the formation of PRPS NCs in the AD brain. Materials and Methods Participant selection Subsequent to approval by the Ottawa Health Science Network Research Ethics Board, tissue blocks of formalin-fixed, paraffin-embedded post-mortem human hippocampus and adjacent parahippocampal gyrus were obtained from the tissue archive in the Division of Anatomical Pathology of the Ottawa Hospital. The cohort included 177 participants including 82 neurologically cognitively unimpaired controls, 30 with advanced AD, 17 with LBD (11 diffuse neocortical, 4 limbic/transitional, 2 brainstem), 14 with primary tauopathy (11 with progressive supranuclear palsy (PSP) and 3 with corticobasal degeneration (CBD)), and an additional 6 cognitively intact participants with significant beta-amyloid deposition. Demographic details regarding the participant cohort as well as the raw data are provided in Supplementary Table 1. Also included in the Table are data indicating the presence or absence of phosphorylated Tau (pTau) neurofibrillary pathology and beta-amyloid plaques in each tissue core. Subjects were included based on retrospective review of post-mortem neuropathology reports. All post-mortem neuropathological diagnoses were performed by a neuropathologist (JW) according to current diagnostic and staging criteria [4, 18, 36, 47]. Specifically, AD neuropathologic change (ADNC) was diagnosed based on the combined analysis of cortical beta amyloid burden according to the Thal staging scheme [60], Braak neurofibrillary stage [9], and cortical neuritic plaque density following Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) criteria [45] to arrive at an "ABC" score as per The National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease [47] (see Supplementary Table 1). A proportion of cases were diagnosed prior to publication of these guidelines and only those receiving a diagnosis of "severe" AD or a Braak score of V or VI were included among the AD cohort. A proportion of cognitively normal controls were assessed for ADNC and only those with "mild" changes were included. Tissue microarray preparation and immunostaining From each of the paraffin blocks of hippocampus and mesial temporal cortex, tissue microarrays (TMAs) were prepared using 2 mm punches centered on the temporal cortex at the depth of the collateral sulcus. This area was selected because it consistently displays NCs in control brains and because it was the area we analysed in our previous study [65]. TMAs were sectioned at a thickness of 5 μm and mounted onto coated slides. Sections were deparaffinized and pre-treated using heat mediated antigen retrieval with EDTA buffer (pH 9.0). Slides were then rehydrated in 1X TBST buffer and blocked for 30 min with Sniper (#Biocare BS966L). Sections were stained using dual labelling immunofluorescence by incubating overnight at 4 degrees in a cocktail of antibodies against PRPS (rabbit polyclonal; 1:100, ProteinTech 15549-1-AP) and the neuronal marker microtubule-associated protein 2 (MAP2; mouse monoclonal; Clone B-8; Santa Cruz Biotechnology sc-74420) or pTau (mouse monoclonal; 1:100, ThermoFisher #MN1020). Sections were washed with 1X TBST and then incubated with goat anti-mouse 594 (#A11005, Invitrogen) and donkey anti-rabbit 488 (#A21206) secondary antibodies for 2 h in the dark at room temperature. This was followed by incubation with a quencher (Vector TrueView Autofluorescence Quenching Kit #SP-8400, Vector Labs) to decrease autofluorescence. Sections were washed, incubated with 5 ug/ml of DAPI (ThermoScientific #62248) and coverslipped. Image and data analysis Images were acquired using a Zeiss AxioImager M2 microscope. To quantify NCs, PRPS/MAP2 stained TMAs were scanned using a Zeiss M1 Slide Scanner. Image analysis was performed on digital images using Highplex FL module in Halo software as well as Zeiss Zen Blue software. The number of MAP2-immunoreactive cells containing NCs in each core was counted manually. Cases with less than 10 neurons per core were eliminated. All statistical analyses for multiple group comparisons were conducted using one-way ANOVA followed by Bonferroni’s multiple comparisons test at α = 0.05 significance. Percentage of NCs present in aggregate with NFTs was determined by manual count. Chi-square test at α = 0.05 significance was used to determine whether there was any association between the presence of NCs and NFTs. To assess beta-amyloid pathology in each core, the TMAs were stained using immunohistochemistry for the detection of beta-amyloid (rabbit polyclonal anti-beta-amyloid; 1:400, Novus NBP2-13075). Staining was performed using the Leica Bond™ system. TMAs were pre-treated using heat mediated antigen retrieval with EDTA buffer (pH 9.0, epitope retrieval solution 2) for 20 minutes. The TMAs were then incubated in primary antibody for 30 minutes at room temperature and detected using an HRP conjugated compact polymer system. Slides were then stained using DAB as the chromogen, counterstained with hematoxylin, mounted and coverslipped. Results PRPS NCs are present in the brain Figure 1 shows PRPS-immunoreactive NCs in double immunostained sections of the punches of temporal cortex in the depth of the collateral sulcus. In both control and disease cases, these were most prevalent in layers II, V, and VI. They were present in both pyramidal neurons as well as non-pyramidal neurons of various morphologies. They showed a variety of shapes, ranging from rod- or thread-like linear or curvilinear structures (Fig. 1a) to dot-shaped structures or large, irregular "crystalloid" sheets (Fig. 1b). Some of the latter showed a rhomboid shape as reported for IMPDH NCs in substantia nigra neurons [64]. In a proportion of neurons, the nucleolus showed intense staining for PRPS (Fig. 1c). This was seen both in neurons with and without NCs. In addition to NCs, most neurons showed innumerable micron-sized curvilinear cytoplasmic threads filling the perikaryal and neuritic cytoplasm and contributing to a punctate staining pattern of the background neuropil (Fig. 1d).
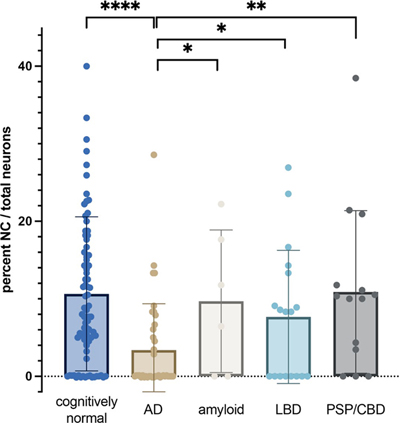
Fig. 1 Dual-labelling immunofluorescence for PRPS (green) and MAP2 (red) in a core of temporal cortex from a control subject. a, b) Neurons containing linear or curvilinear (a) or irregular sheet-like (b) NCs. c) PRPS staining of the nucleoli in two neurons. In both cases, there are small NCs juxtaposed to the nucleoli (arrowheads). d) Higher magnification of the proximal apical dendrite of the pyramidal neuron in b showing small curvilinear cytoplasmic PRPS filaments within the dendrite and dot-like structures throughout the surrounding neuropil. Scale bars = 20 microns. PRPS NCs are reduced in the AD temporal cortex The box plot in Fig. 2 shows the results of our quantitative analysis. There was significant inter-individual variability in the density of NCs. Quantitative analysis confirmed a significant depletion of NCs in participants with AD relative to cognitively intact age-matched controls, including those with heavy beta-amyloid deposition, as well as those with LBD and 4R tauopathies (PSP/CBD) (ANOVA p < 0.02, see Figure 2 for pairwise comparisons). Patients with LBD and 4-repeat tauopathies did not differ from controls (p = 0.2749 and 0.9897, respectively). The presence of heavy beta-amyloid deposition in cognitively normal individuals did not impact NC density relative to the control population (p = 0.9322). There were no discernible qualitative differences in the appearance or morphology of NCs between AD participants, any other disease groups, and controls.
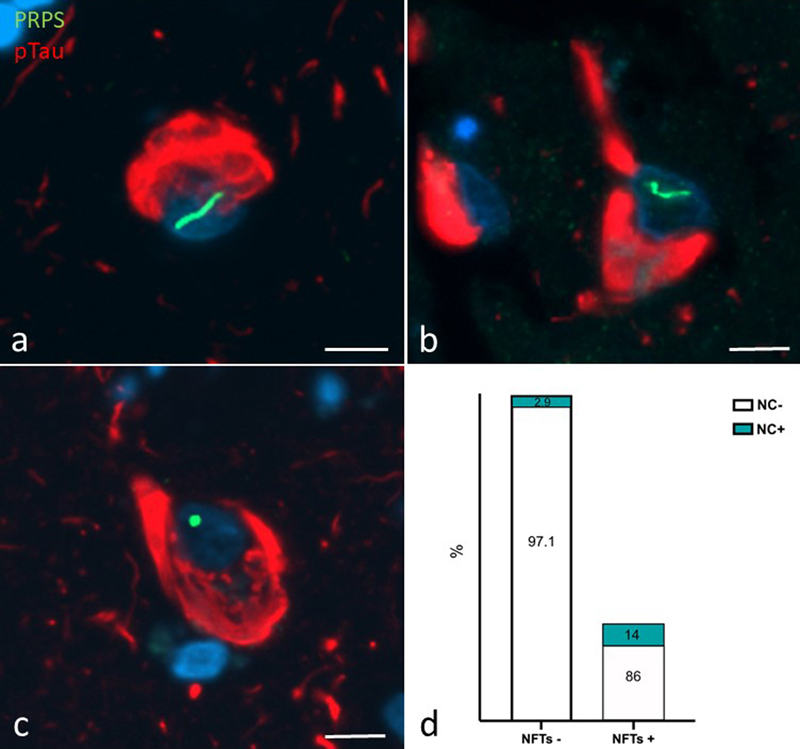
Fig. 2 Box plot comparing frequency of NCs in cores of temporal cortex from our cohort. ANOVA for the entire group p < 0.02. Pairwise comparisons conducted using Bonferroni’s multiple comparisons test. Comparisons reaching the significance level (p < 0.05) are shown on the graph. All other comparisons did not reach this level. AD; Alzheimer's disease, amyloid; cognitively normal subjects with high density of amyloid plaques, LBD; Lewy body disease, PSP/CBD; progressive supranuclear palsy/corticobasal degeneration. ****p < 0.0001, *p < 0.05., **p < 0.005. PRPS NCs are more common in NFT-bearing neurons We explored a possible relationship between NCs and neurofibrillary tangles (NFTs) using double immunofluorescence staining of the TMAs for PRPS and pTau (Fig. 3a–c). A significantly higher proportion of NFT-bearing neurons contained NCs relative to NFT-negative neurons, suggesting an association between these two proteinaceous bodies. Specifically, of 64 NFT-containing neurons, 9 (14 %) contained NCs. Conversely, of 4,977 neurons lacking NFTs, only 145 (2.9 %) contained NCs (Fig. 3d; Chi-squared p < 0.00001).
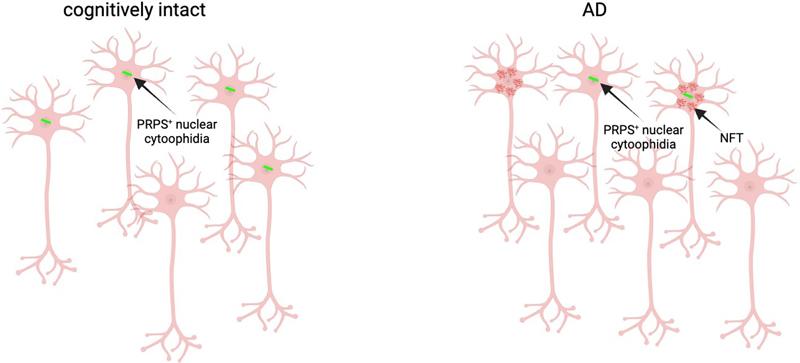
Fig. 3 a–c) Dual labelling immunofluorescence for PRPS (green) and pTau (red) showing curvilinear (a), thread-like (b) and dot-like (c) NCs in tangle-bearing neurons. Scale bars = 10 microns. d) Bar graph showing percentage of NFT-positive and NFT-negative neurons with (blue) and without (white) NCs. Chi-square test shows a significantly higher proportion of NFT bearing neurons contain NCs (p < 0.00001). Discussion Here we demonstrate a significant depletion of intranuclear cytoophidia comprised of the key nucleotide-synthesizing enzyme PRPS from neurons in the AD cerebral cortex, implicating these enigmatic structures in the pathogenesis of AD. This study confirms and extends the results of our previous study demonstrating a significant decrement in the density of intranuclear rodlets (INRs) in the temporal cortex of individuals with AD relative to cognitively intact controls and those with dementia with Lewy bodies [65]. These structures correspond to the "rodlets of Roncoroni" described by the classical microscopists (see [63]). The functional significance of these structures has remained enigmatic. The demonstration that they are immunoreactive for the nucleotide synthesizing enzymes PRPS provides a novel functional context; linking them to the bundles of metabolic enzyme filaments known as "cytoophidia" recently described by several authors (for a review, see [11]). Interestingly, we have demonstrated that the PRPS NCs described in this study are also immunoreactive for the rate limiting enzyme in purine nucleotide synthesis, IMPDH [63]. Co-localization of metabolic enzymes in cytoophidia is common. For example, a similar interaction between IMPDH and the pyrimidine-synthesizing enzyme cytidine triphosphate synthase (CTPS) in cytoophidia has been implicated as a substrate to co-ordinate the cross-regulation of purine and pyrimidine nucleotide synthesis [14]. In a screen of 440 yeast metabolic enzymes, Noree et al. showed the assembly of multiple metabolic enzymes into rod-shaped structures [50]. Interestingly, enzymes that co-assembled, like IMPDH and PRPS, were those that acted at branch points within the de novo purine biosynthetic pathway, further suggesting that this co-localization reflects some form of regulatory mechanism in the nucleotide biosynthetic pathway. The majority of mechanistic studies have linked cytoophidia formation to the dynamic regulation of enzyme activity and thus, in the case of PRPS (with IMPDH), to the synthesis of purine nucleotides. Purine nucleotides are essential for the proper development and maintenance of the brain. Guanine-based purines, and especially GTP and its breakdown product guanosine, have neuromodulatory properties (reviewed in [59]) with neuroprotective, neurotrophic, and neuritogenic capacity [23, 24, 49, 59]. These are mediated through intracellular modulation of G-proteins as well as via extracellular signaling by purines themselves [59]. GTP plays a critical role in tubulin polymerization for microtubule growth [13] and is a substrate for the multitude of small GTPases involved in neurite growth and synaptogenesis [19, 61]. Our findings are consistent with evidence for the involvement of nucleotide-synthesizing enzymes, and possibly their filamentation, in neurological diseases [27]. Loss-of-function mutations in PRPS1 (encoded on the X-chromosome), culminate in a constellation of neurodevelopmental symptoms in males referred to eponymously as Arts syndrome and encompassing sensorineural hearing loss, hypotonia, ataxia, developmental delay, and intellectual disability [16, 17]. Similarly, mutations in IMPDH2 result in variable combinations of intellectual disability, speech impairment, hand tremor, gait instability, and dystonia [51, 52, 66]. Why mutations in these enzymes have a relatively selective impact on the nervous system despite their widespread systemic expression has not been adequately explained. The relatively selective localization of NCs to the brain despite the widespread expression of their constituent enzymes is provocative in this regard. Intriguingly, a proportion of the disease-causing mutations in both PRPS1, as well as IMPDH alter filament assembly [25, 52], suggesting a beneficial role for filament formation. Consistent with this, Flores-Mendez and co-workers recently demonstrated a neuroprotective role for IMPDH2 filament formation in neurodegeneration [21]. Understanding the mechanisms through which NCs might contribute to AD pathogenesis is predicated on elucidating their physiological significance in neurons, which is currently unknown. A canonical role for PRPS filaments in regulating enzyme activity makes sense in the cytoplasmic compartment where nucleotides are synthesized. Indeed, we postulate that the small cytoplasmic filaments we describe herein are engaged in this, perhaps in association with mitochondria (see [64]), by analogy with purinosomes [22]. On the other hand, the functional significance of PRPS1 filament assemblies within the nuclear compartment of neurons remains to be explored. There is evidence that PRPS, as well as IMPDH, can localize to the nucleus [32, 37]. We have described the presence of IMPDH NCs within the nuclei of substantia nigra neurons where they display marked age-associated morphological alterations [64]. PRPS1-immunoreactive rods have also been described in the nucleus of neurons in vitro [50] and in vivo [63]. This is consistent with evidence for a nuclear localization and function of a growing number of metabolic enzymes [7]. We propose two non-mutually exclusive possibilities regarding the significance of PRPS/IMPDH rods in the nuclear compartment. First, it is conceivable that NCs regulate the synthesis of purines within the nucleus, providing a proximate, "hyperlocal" source of nucleotides at loci of high nucleotide demand. Indeed, previous reports have shown that IMPDH localization is responsive to GTP demand and that this localization enhances de novo GTP synthesis [2, 6, 15, 33, 62, 67]. This model might explain the common spatial association of NCs with the nucleolus which consumes a vast quantity of purine nucleotides for the synthesis of rRNA [34, 35]. By analogy, there is evidence that acetyl-CoA, essential for histone acetylation, is synthesized in the nucleus by the metabolic enzymes ATP-citrate lyase (ACLY), acyl-CoA synthetase short chain family member 2 (ACSS2) and the pyruvate dehydrogenase complex (PDC) [8]. It has been hypothesized that these enzymes localize to nuclear foci and generate acetyl CoA at sites of histone acetyltransferase activity. A role for NCs in the generation of GTP in the nuclear compartment is predicated on the local presence of downstream enzymes in this pathway to facilitate metabolic channeling. Indeed, GMPS, which generates GMP from IMPDH-generated XMP, has been described in the nucleus [56]. Alternatively, there is increasing evidence that metabolic enzymes engage in moonlighting, non-enzymatic activities in the nucleus [8]. For example, in Saccharomyces cerevesiae, the nuclear localization of PRPS isoforms has been implicated in the maintenance of cell wall integrity [57]. In Drosophila, IMPDH enters the nucleus in response to oxidative and/or replicative stress, binds single-stranded CT-rich regulatory DNA elements and functions as a transcriptional repressor [37]. In breast cancer cells, chromatin-bound IMPDH interacts with PARP1 and has been implicated in DNA repair by regulating the availability of NAD+ [20]. It is tempting to speculate that neurons exploit the capacity of PRPS and IMPDH to form filaments as a mechanism to regulate the nucleoplasmic availability of these enzymes to perform these moonlighting functions. According to the beta-amyloid hypothesis of AD pathogenesis, the natural history of AD is characterized by a protracted prodromal period, lasting at least a decade, during which beta amyloid accumulates in cortical plaques without overt physiological or cognitive sequelae. At some point, amyloid is thought to catalyze the expansion of neuronal tau pathology from the basal temporal cortex, (where NFTs remain confined in the normal elderly), to the neocortex, and trigger the development of dementia. Our study demonstrates no definite relationship between NC loss and beta-amyloid load. The dissociation of NC loss from beta amyloid deposition is consistent with evidence that nucleotide dysmetabolism in the AD brain occurs independently of ß-amyloid plaques [1, 3, 26, 29–31]. On the other hand, our results suggest that NC loss occurs subsequent to beta-amyloid deposition, and, based on the amyloid hypothesis, at the stage of NFT expansion. This is a critical phase of the AD pathogenetic cascade as NFT formation correlates best with cognitive decline. Notably, despite a global loss of NCs from the AD brain, our results indicate that remaining NCs tend to associate with NFT-bearing neurons. This apparent contradiction may relate to the fact that NFT-bearing neurons comprise a relatively small proportion of the total neuronal population in the AD cortex (1.28 % in our cohort). Thus, assuming that NC loss is independent of beta-amyloid and tau deposition, the net quantitative effect is a global loss of NCs, despite their preferential localization to NFT-bearing neurons. Why remaining NCs tend to form in NFT-bearing neurons remains to be explored. It is tempting to speculate that NFT formation imposes a metabolic burden on the neuron culminating in NC formation. This would be consistent with our previous study demonstrating increased intranuclear rods in the substantia nigra in response to the neurotoxin MPTP [39]. Conversely, it is possible that the metabolic status of the neuron which results in NC formation predisposes neurons to tau hyperphosphorylation and NFT pathology. Elucidating the mechanistic substrates underlying this association may lead to novel pathogenetic concepts and therapeutic targets. Our results may also have disease implications in a broader context. For example, NCs were not significantly reduced in frequency in the cortex of LBD subjects. This supports the status of NC depletion as a disease-specific biomarker for AD as opposed to LBD. In addition, we have demonstrated the presence of INRs in insulin-secreting pancreatic beta cells [55]. Like AD, we have demonstrated a significant depletion of INRs from beta cells in human type 2 diabetes [68] as well as in animal models of diabetes [44]. This is intriguing in light of evidence for: 1) a shared pathogenetic mechanism underlying AD and type 2 diabetes [46] and 2) an important role for purine nucleotide metabolism in beta cell function and survival and in diabetes [43]. Moreover, we have demonstrated evidence for an inverse correlation between cellular secretory activity and INR formation [63]. Notably, beta-cell and neuronal hyperactivity are hallmarks of diabetes and AD [10], providing an additional, or alternative unifying explanation for NC depletion in the two diseases. NC depletion may represent a morphological signpost indicating a common metabolic substrate contributing to the pathogenesis of these two related disorders of ageing. Our study has some limitations. Participant recruitment was based on retrospective review of neuropathology reports. A small proportion of the AD cases were diagnosed prior to the currently recommended diagnostic criteria [47], precluding assignment of an "ABC" score. Only cases with "severe" ADNC or a Braak score of V or VI were included in the AD cohort. The objective of this study was to confirm our previous findings in the context of new knowledge regarding the possible functional significance of, and novel marker for, these intranuclear structures. Consequently, the comparison of subjects with only "high" ADNC with controls is dichotomous. Future studies will provide a more granular understanding of the role of NCs in the AD pathogenic cascade by analyzing their status at "low" and "intermediate" ADNC stages. This is being pursued in mouse studies as well. There is increasing recognition that TDP-43 proteinopathy in the form of limbic-predominant age-related TDP-43 encephalopathy (LATE) is a common co-pathology in ADNC. We did not examine the impact of this co-pathology on NC status in the present study. n summary, we have demonstrated: 1) that neuronal PRPS NCs are reduced in frequency in the AD temporal cortex and 2) that remaining NCs show an association with NFT-containing neurons (Fig. 4). The results of this study provide an unexplored avenue for investigation with respect to AD pathogenesis. The potential involvement of PRPS NCs in the cellular mechanisms underlying AD-associated neurodegeneration is consistent with evidence for metabolic dysfunction in AD. Future mechanistic studies addressing the functional significance of PRPS NCs will elucidate their role in healthy neurons and in disease. If these confirm that intranuclear filamentation of these metabolic enzymes plays a role in neuronal health and metabolism, as has been suggested, its failure in AD may provide a novel, biologically rational therapeutic target.
Fig. 4 Schematic summary of the results of the present study. Neuronal NCs (green) are reduced in frequency in the brains of AD participants. Remaining NCs are relatively more frequent in NFT (red)-bearing neurons. Declarations Author Contributions Conceptualization: John Woulfe, David Munoz; Methodology: John Woulfe, Alyona Ivanova; Formal analysis and Investigation: John Woulfe, Alyona Ivanova; Writing-original draft preparation: John Woulfe; Writing-review and editing: David Munoz, Alyona Ivanova; Resources: John Woulfe, David Munoz; Supervision: John Woulfe, David Munoz. Funding statement No funding was received for conducting this study. Conflict of interest statement The authors have no relevant financial or non-financial interests to disclose. Ethics Approval This retrospective post-mortem study was conducted in accordance with the ethical standards of The Ottawa Hospital. Data Availability The data is available from the corresponding author upon reasonable request. Raw data is available in Supplementary Table 1. Acknowledgements The authors would like to acknowledge the excellent assistance of Dr. Sharlene Faulkes, Marjan Khalili Mahani, and Zaida Ticas and the Louise Pelletier Histology Core Facility (RRID: SCR_021737), Department of Pathology and Laboratory Medicine, University of Ottawa. References 1. Alonso-Andres P, Albasanz JL, Ferrer I, Martin M (2018) Purine-related metabolites and their converting enzymes are altered in frontal, parietal and temporal cortex at early stages of Alzheimer's disease pathology. Brain Pathol 28: 933-946. https://doi.org/10.1111/bpa.12592 2. An S, Kumar R, Sheets ED, Benkovic SJ (2008) Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science 320: 103-106. https://doi.org/10.1126/science.1152241 3. Ansoleaga B, Jove M, Schluter A, Garcia-Esparcia P, Moreno J, Pujol A, Pamplona R, Portero-Otin M, Ferrer I (2015) Deregulation of purine metabolism in Alzheimer's disease. Neurobiol Aging 36: 68-80. https://doi.org/10.1016/j.neurobiolaging.2014.08.004 4. Attems J, Toledo JB, Walker L, Gelpi E, Gentleman S, Halliday G, Hortobagyi T, Jellinger K, Kovacs GG, Lee EBet al (2021) Neuropathological consensus criteria for the evaluation of Lewy pathology in post-mortem brains: a multi-centre study. Acta Neuropathol 141: 159-172. https://doi.org/10.1007/s00401-020-02255-2 5. Aughey GN, Liu JL (2015) Metabolic regulation via enzyme filamentation. Crit Rev Biochem Mol Biol 51: 282-293. https://doi.org/10.3109/10409238.2016.1172555 6. Bianchi G, Longhi S, Grandori R, Brocca S (2020) Relevance of Electrostatic Charges in Compactness, Aggregation, and Phase Separation of Intrinsically Disordered Proteins. Int J Mol Sci 21:. https://doi.org/10.3390/ijms21176208 7. Boon R, Silveira GG, Mostoslavsky R (2020) Nuclear metabolism and the regulation of the epigenome. Nat Metab 2: 1190-1203. https://doi.org/10.1038/s42255-020-00285-4 8. Boukouris AE, Zervopoulos SD, Michelakis ED (2016) Metabolic Enzymes Moonlighting in the Nucleus: Metabolic Regulation of Gene Transcription. Trends Biochem Sci 41: 712-730. https://doi.org/10.1016/j.tibs.2016.05.013 9. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112: 389-404. https://doi.org/10.1007/s00401-006-0127-z 10. Busche MA, Konnerth A (2015) Neuronal hyperactivity--A key defect in Alzheimer's disease? BioEssays 37: 624-632. https://doi.org/10.1002/bies.201500004 11. Calise SJ, Bizzaro N, Nguyen T, Bassetti D, Porcelli B, Almi P, Barberio G, Pesce G, Satoh M, Chan EK (2016) Anti-rods/rings autoantibody seropositivity does not affect response to telaprevir treatment for chronic hepatitis C infection. Auto Immun Highlights 7: 15. https://doi.org/10.1007/s13317-016-0087-9 12. Calise SJ, Chan EKL (2020) Anti-rods/rings autoantibody and IMPDH filaments: an update after fifteen years of discovery. Autoimmun Rev 19: 102643. https://doi.org/10.1016/j.autrev.2020.102643 13. Carlier MF (1982) Guanosine-5'-triphosphate hydrolysis and tubulin polymerization. Review article. Mol Cell Biochem 47: 97-113. https://doi.org/10.1007/BF00234410 14. Chang CC, Keppeke GD, Sung LY, Liu JL (2018) Interfilament interaction between IMPDH and CTPS cytoophidia. FEBS J 285: 3753-3768. https://doi.org/10.1111/febs.14624 15. Chang CC, Lin WC, Pai LM, Lee HS, Wu SC, Ding ST, Liu JL, Sung LY (2015) Cytoophidium assembly reflects upregulation of IMPDH activity. J Cell Sci 128: 3550-3555. https://doi.org/10.1242/jcs.175265 16. de Brouwer AP, van Bokhoven H, Nabuurs SB, Arts WF, Christodoulou J, Duley J (2010) PRPS1 mutations: four distinct syndromes and potential treatment. Am J Hum Genet 86: 506-518. https://doi.org/10.1016/j.ajhg.2010.02.024 17. de Brouwer AP, Williams KL, Duley JA, van Kuilenburg AB, Nabuurs SB, Egmont-Petersen M, Lugtenberg D, Zoetekouw L, Banning MJ, Roeffen Met al (2007) Arts syndrome is caused by loss-of-function mutations in PRPS1. Am J Hum Genet 81: 507-518. https://doi.org/10.1086/520706 18. Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, Jellinger K, Lantos PL, Lippa CF, Mirra SSet al (2002) Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 61: 935-946. https://doi.org/10.1093/jnen/61.11.935 19. Duman JG, Mulherkar S, Tu YK, J XC, Tolias KF (2015) Mechanisms for spatiotemporal regulation of Rho-GTPase signaling at synapses. Neurosci Lett 601: 4-10. https://doi.org/10.1016/j.neulet.2015.05.034 20. Espinar L, Garcia-Cao M, Schmidt A, Kourtis S, Ganez Zapater A, Aranda-Vallejo C, Ghose R, Garcia-Lopez L, Sheraj I, Pardo-Lorente Net al (2024) Nuclear IMPDH2 controls the DNA damage response by modulating PARP1 activity. Nature communications 15: 9515. https://doi.org/10.1038/s41467-024-53877-z 21. Flores-Mendez M, Ohl L, Roule T, Zhou Y, Tintos-Hernandez JA, Walsh K, Ortiz-Gonzalez XR, Akizu N (2024) IMPDH2 filaments protect from neurodegeneration in AMPD2 deficiency. EMBO Rep 25: 3990 - 4012:. https://doi.org/10.1038/s44319-024-00218-2 22. French JB, Jones SA, Deng H, Pedley AM, Kim D, Chan CY, Hu H, Pugh RJ, Zhao H, Zhang Yet al (2016) Spatial colocalization and functional link of purinosomes with mitochondria. Science 351: 733-737. https://doi.org/10.1126/science.aac6054 23. Gysbers JW, Rathbone MP (1992) Guanosine enhances NGF-stimulated neurite outgrowth in PC12 cells. Neuroreport 3: 997-1000. https://doi.org/10.1097/00001756-199211000-00013 24. Gysbers JW, Rathbone MP (1996) Neurite outgrowth in PC12 cells is enhanced by guanosine through both cAMP-dependent and -independent mechanisms. Neurosci Lett 220: 175-178. https://doi.org/10.1016/s0304-3940(96)13253-5 25. Hvorecny KL, Hargett K, Quispe JD, Kollman JM (2023) Human PRPS1 filaments stabilize allosteric sites to regulate activity. Nat Struct Mol Biol 30: 391-402. https://doi.org/10.1038/s41594-023-00921-z 26. Isobe C, Abe T, Terayama Y (2010) Levels of reduced and oxidized coenzyme Q-10 and 8-hydroxy-2'-deoxyguanosine in the CSF of patients with Alzheimer's disease demonstrate that mitochondrial oxidative damage and/or oxidative DNA damage contributes to the neurodegenerative process. J Neurol 257: 399-404. https://doi.org/10.1007/s00415-009-5333-x 27. Jinnah HA, Sabina RL, Van Den Berghe G (2013) Metabolic disorders of purine metabolism affecting the nervous system. Handb Clin Neurol 113: 1827-1836. https://doi.org/10.1016/B978-0-444-59565-2.00052-6 28. Johnson MC, Kollman JM (2020) Cryo-EM structures demonstrate human IMPDH2 filament assembly tunes allosteric regulation. eLife 9:. https://doi.org/10.7554/eLife.53243 29. Jove M, Portero-Otin M, Naudi A, Ferrer I, Pamplona R (2014) Metabolomics of human brain aging and age-related neurodegenerative diseases. J Neuropathol Exp Neurol 73: 640-657. https://doi.org/10.1097/NEN.0000000000000091 30. Kaddurah-Daouk R, Rozen S, Matson W, Han X, Hulette CM, Burke JR, Doraiswamy PM, Welsh-Bohmer KA (2011) Metabolomic changes in autopsy-confirmed Alzheimer's disease. Alzheimers Dement 7: 309-317. https://doi.org/10.1016/j.jalz.2010.06.001 31. Kaddurah-Daouk R, Zhu H, Sharma S, Bogdanov M, Rozen SG, Matson W, Oki NO, Motsinger-Reif AA, Churchill E, Lei Zet al (2013) Alterations in metabolic pathways and networks in Alzheimer's disease. Transl Psychiatry 3: e244. https://doi.org/10.1038/tp.2013.18 32. Kaida A, Ariumi Y, Baba K, Matsubae M, Takao T, Shimotohno K (2005) Identification of a novel p300-specific-associating protein, PRS1 (phosphoribosylpyrophosphate synthetase subunit 1). Biochem J. 391: 239-247. https://doi.org/10.1042/BJ20041308 33. Keppeke GD, Chang CC, Peng M, Chen LY, Lin WC, Pai LM, Andrade LEC, Sung LY, Liu JL (2018) IMP/GTP balance modulates cytoophidium assembly and IMPDH activity. Cell Div 13: 5. https://doi.org/10.1186/s13008-018-0038-0 34. Kofuji S, Hirayama A, Eberhardt AO, Kawaguchi R, Sugiura Y, Sampetrean O, Ikeda Y, Warren M, Sakamoto N, Kitahara Set al (2019) IMP dehydrogenase-2 drives aberrant nucleolar activity and promotes tumorigenesis in glioblastoma. Nat Cell biol 21: 1003-1014. https://doi.org/10.1038/s41556-019-0363-9 35. Kofuji S, Sasaki AT (2020) GTP metabolic reprogramming by IMPDH2: unlocking cancer cells' fuelling mechanism. J Biochem 168: 319-328. https://doi.org/10.1093/jb/mvaa085 36. Kovacs GG, Lukic MJ, Irwin DJ, Arzberger T, Respondek G, Lee EB, Coughlin D, Giese A, Grossman M, Kurz Cet al (2020) Distribution patterns of tau pathology in progressive supranuclear palsy. Acta Neuropathol 140: 99-119. https://doi.org/10.1007/s00401-020-02158-2 37. Kozhevnikova EN, van der Knaap JA, Pindyurin AV, Ozgur Z, van Ijcken WF, Moshkin YM, Verrijzer CP (2012) Metabolic enzyme IMPDH is also a transcription factor regulated by cellular state. Mol Cell 47: 133-139. https://doi.org/10.1016/j.molcel.2012.04.030 38. Kuukasjarvi A, Landoni JC, Kaukonen J, Juhakoski M, Auranen M, Torkkeli T, Velagapudi V, Suomalainen A (2021) IMPDH2: a new gene associated with dominant juvenile-onset dystonia-tremor disorder. Eur J Hum Genet 29: 1833-1837. https://doi.org/10.1038/s41431-021-00939-1 39. Lamba W, Prichett W, Munoz D, Park DS, Woulfe JM (2005) MPTP induces intranuclear rodlet formation in midbrain dopaminergic neurons. Brain Res 1066: 86-91. https://doi.org/10.1016/j.brainres.2005.10.058 40. Liu JL (2016) The Cytoophidium and Its Kind: Filamentation and Compartmentation of Metabolic Enzymes. Annu Rev Cell Dev Biol 32: 349-372. https://doi.org/10.1146/annurev-cellbio-111315-124907 41. Lynch EM, Kollman JM, Webb BA (2020) Filament formation by metabolic enzymes-A new twist on regulation. Curr Opin Cell Biol 66: 28-33. https://doi.org/10.1016/j.ceb.2020.04.006 42. Mercati O, Abi Warde MT, Lina-Granade G, Rio M, Heide S, de Lonlay P, Ceballos-Picot I, Robert MP, Couloigner V, Beltrand Jet al (2020) PRPS1 loss-of-function variants, from isolated hearing loss to severe congenital encephalopathy: New cases and literature review. Eur J Med Genet 63: 104033. https://doi.org/10.1016/j.ejmg.2020.104033 43. Metz SA, Kowluru A (1999) Inosine monophosphate dehydrogenase: A molecular switch integrating pleiotropic GTP-dependent beta-cell functions. Proc Assoc Am Physicians 111: 335-346. https://doi.org/10.1046/j.1525-1381.1999.99245.x 44. Milman P, Fu A, Screaton RA, Woulfe JM (2010) Depletion of intranuclear rodlets in mouse models of diabetes. Endocrine pathology 21: 230-235. https://doi.org/10.1007/s12022-010-9136-5 45. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L (1991) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41: 479-486. https://doi.org/10.1212/wnl.41.4.479 46. Mittal K, Katare DP (2016) Shared links between type 2 diabetes mellitus and Alzheimer's disease: A review. Diabetes Metab Syndr 10: S144-149. https://doi.org/10.1016/j.dsx.2016.01.021 47. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SSet al (2012) National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 123: 1-11. https://doi.org/10.1007/s00401-011-0910-3 48. Natsumeda Y, Ohno S, Kawasaki H, Konno Y, Weber G, Suzuki K (1990) Two distinct cDNAs for human IMP dehydrogenase. J Biol Chem 265: 5292-5295. https://doi.org/10.1016/S0021-9258(19)34120-1 49. Neary JT, Rathbone MP, Cattabeni F, Abbracchio MP, Burnstock G (1996) Trophic actions of extracellular nucleotides and nucleosides on glial and neuronal cells. Trends Neurosci 19: 13-18. https://doi.org/10.1016/0166-2236(96)81861-3 50. Noree C, Begovich K, Samilo D, Broyer R, Monfort E, Wilhelm JE (2019) A quantitative screen for metabolic enzyme structures reveals patterns of assembly across the yeast metabolic network. Mol Biol Cell 30: 2721-2736. https://doi.org/10.1091/mbc.E19-04-0224 51. O'Neill AG, Burrell AL, Zech M, Elpeleg O, Harel T, Edvardson S, Mor-Shaked H, Rippert AL, Nomakuchi T, Izumi Ket al (2023) Neurodevelopmental disorder mutations in the purine biosynthetic enzyme IMPDH2 disrupt its allosteric regulation. J Biol Chem 299: 105012. https://doi.org/10.1016/j.jbc.2023.105012 52. O'Neill AG, Burrell AL, Zech M, Elpeleg O, Harel T, Edvardson S, Shaked HM, Rippert AL, Nomakuchi T, Izumi Ket al (2023) Point mutations in IMPDH2 which cause early-onset neurodevelopmental disorders disrupt enzyme regulation and filament structure. bioRxiv:. https://doi.org/10.1101/2023.03.15.532669 53. Pareek V, Tian H, Winograd N, Benkovic SJ (2020) Metabolomics and mass spectrometry imaging reveal channeled de novo purine synthesis in cells. Science 368: 283-290. https://doi.org/10.1126/science.aaz6465 54. Pedley AM, Pareek V, Benkovic SJ (2022) The Purinosome: A Case Study for a Mammalian Metabolon. Annu Rev Biochem 91: 89-106. https://doi.org/10.1146/annurev-biochem-032620-105728 55. Prichett W, Milman P, Gagnon J, Munoz DG, Woulfe J (2007) Intranuclear rodlets in human pancreatic islet cells. Pancreas 35: 207-211. https://doi.org/10.1097/MPA.0b013e318067fee1 56. Reddy BA, van der Knaap JA, Bot AG, Mohd-Sarip A, Dekkers DH, Timmermans MA, Martens JW, Demmers JA, Verrijzer CP (2014) Nucleotide biosynthetic enzyme GMP synthase is a TRIM21-controlled relay of p53 stabilization. Mol Cell 53: 458-470. https://doi.org/10.1016/j.molcel.2013.12.017 57. Schneiter R, Carter AT, Hernando Y, Zellnig G, Schweizer LM, Schweizer M (2000) The importance of the five phosphoribosyl-pyrophosphate synthetase (Prs) gene products of Saccharomyces cerevisiae in the maintenance of cell integrity and the subcellular localization of Prs1p. Microbiology (Reading) 146 Pt 12: 3269-3278. https://doi.org/10.1099/00221287-146-12-3269 58. Simonet JC, Burrell AL, Kollman JM, Peterson JR (2020) Freedom of assembly: metabolic enzymes come together. Mol Biol Cell 31: 1201-1205. https://doi.org/10.1091/mbc.E18-10-0675 59. Tasca CI, Lanznaster D, Oliveira KA, Fernandez-Duenas V, Ciruela F (2018) Neuromodulatory Effects of Guanine-Based Purines in Health and Disease. Front Cell Neurosci 12: 376. https://doi.org/10.3389/fncel.2018.00376 60. Thal DR, Rub U, Orantes M, Braak H (2002) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58: 1791-1800. https://doi.org/10.1212/wnl.58.12.1791 61. Tolias KF, Duman JG, Um K (2011) Control of synapse development and plasticity by Rho GTPase regulatory proteins. Prog Neurobiol 94: 133-148. https://doi.org/10.1016/j.pneurobio.2011.04.011 62. Wolfe K, Kofuji S, Yoshino H, Sasaki M, Okumura K, Sasaki AT (2019) Dynamic compartmentalization of purine nucleotide metabolic enzymes at leading edge in highly motile renal cell carcinoma. Biochem Biophys Res Commun 516: 50-56. https://doi.org/10.1016/j.bbrc.2019.05.190 63. Woulfe J, Munoz D (2024) Roncoroni Re-Visited: The Neuronal Intranuclear Rodlet Comes of Age. J Comp Neurol 532: e25662. https://doi.org/10.1002/cne.25662 64. Woulfe J, Munoz DG, Gray DA, Jinnah HA, Ivanova A (2024) Inosine monophosphate dehydrogenase intranuclear inclusions are markers of aging and neuronal stress in the human substantia nigra. Neurobiol Aging 134: 43-56. https://doi.org/10.1016/j.neurobiolaging.2023.11.005 65. Woulfe JM, Hammond R, Richardson B, Sooriabalan D, Parks W, Rippstein P, Munoz DG (2002) Reduction of neuronal intranuclear rodlets immunoreactive for tubulin and glucocorticoid receptor in Alzheimer's disease. Brain Pathol 12: 300-307. https://doi.org/10.1111/j.1750-3639.2002.tb00444.x 66. Zech M, Jech R, Boesch S, Skorvanek M, Weber S, Wagner M, Zhao C, Jochim A, Necpal J, Dincer Yet al (2020) Monogenic variants in dystonia: an exome-wide sequencing study. Lancet Neurol 19: 908-918. https://doi.org/10.1016/S1474-4422(20)30312-4 67. Zhao H, Chiaro CR, Zhang L, Smith PB, Chan CY, Pedley AM, Pugh RJ, French JB, Patterson AD, Benkovic SJ (2015) Quantitative analysis of purine nucleotides indicates that purinosomes increase de novo purine biosynthesis. J Biol Chem 290: 6705-6713. https://doi.org/10.1074/jbc.M114.628701 68. Zhou YY, El Hallani S, Balaa F, Mohammad W, Gray DA, Woulfe J (2017) Depletion of Beta Cell Intranuclear Rodlets in Human Type II Diabetes. Endocr Pathol 28: 282-286. https://doi.org/10.1007/s12022-017-9499-y
Copyright: © 2025 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License ( https://creativecommons.org/licenses/by/4.0/ ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver ( https://creativecommons.org/publicdomain/zero/1.0/ ) applies to the data made available in this article, unless otherwise stated. |