|
|
|
Free Neuropathology 5:26 (2024) |
|
Review |
|
Neurotrauma: 2024 update |
|
David S. Priemer1,2, Daniel P. Perl1,2 |
|
|
Corresponding author: |
|
Submitted: 09 September 2024 |
|
Keywords: Traumatic brain injury, Chronic traumatic encephalopathy, TDP43, Neurovascular, Intimate partner violence, Blast |
|
Abstract 2023 was an important year for research in traumatic brain injury (TBI), particularly as it concerned interests in neuropathology. After reviewing the literature, we present the advancements that we felt were of particular importance to the neuropathology community. Highlighted are articles that report upon: (1) the first large-cohort assessment for the neuropathology of intimate partner violence, (2) the assessment of chronic traumatic encephalopathy (CTE) in young athletes, (3) the observation of cortical sulcal depth vascular changes in CTE, (4) a proposal for a tau immunohistochemical panel to evaluate complex cases of CTE in the context of multiple tauopathies, (5) the relationship of TBI and/or CTE with TDP-43 pathology, (6) repetitive TBI inducing pathology in C9orf72-transgenic mice, (7) radiologic patterns of head and neck injury following vehicular underbody blast exposure, (8) chronic alterations in brain metal content following repetitive impact TBI, (9) neurovascular unit injury following low-level blast exposure, and finally (10) an assessment of Muhammad Ali’s clinical history leading to the conclusion that he suffered from young-onset, idiopathic Parkinson Disease. We close our writing with in memoriam to Dr. Byron A. Kakulas, a renowned figure in the neuropathology of spinal cord injury who we lost in 2023. |
|
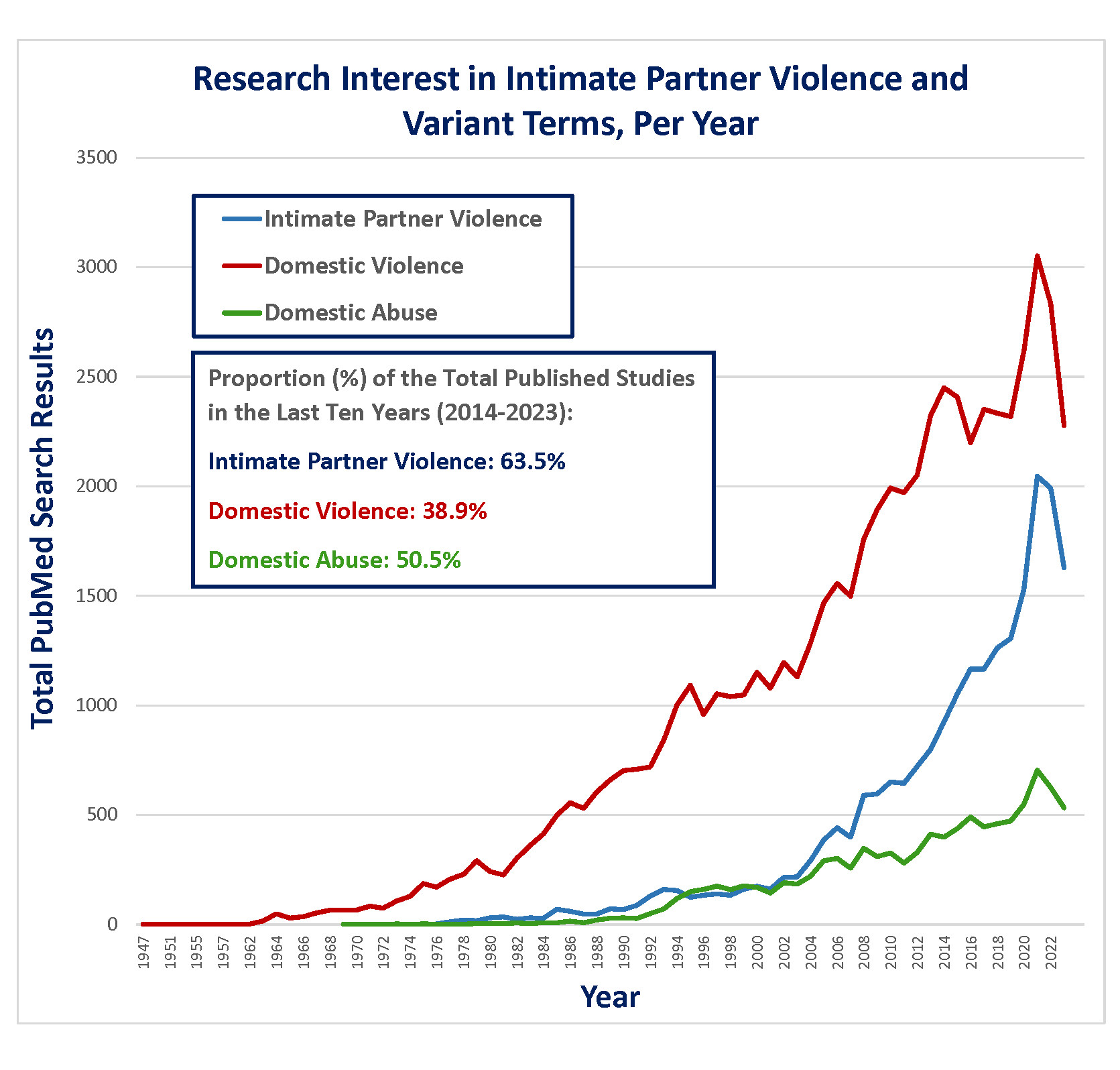
Introduction The year 2023 was an especially fruitful year in the study of traumatic brain injury (TBI), particularly as it concerned aspects of neuropathology, demonstrating that interest in the field remains robust. Many notable studies were published, providing many impactful advancements in knowledge. After conducting a thorough review of the literature regarding neurotrauma over the year of 2023, herein we present and summarize the discoveries that we felt were of particular importance and relevance to the neuropathology community. We conclude our review with memoriam to the life of Dr. Byron A. Kakulas, a pioneer in the neuropathology of spinal cord injury, who died in 2023 at the age of 90. The neuropathology of intimate partner violence While the large focus of research on the chronic neuropathologic effects of repetitive TBI has been dedicated to contact sports athletes and, to a lesser degree, military service, there are other circumstances in which repetitive TBI and its long-term sequelae must be investigated. One important setting is intimate partner violence (IPV), as it is a remarkably common global phenomenon of increasing medical research interest (Figure 1) and as previous, highly-cited anecdotal reports have implicated this exposure with risk of chronic traumatic encephalopathy (CTE).1-3 In 2023, authors Dams-O’Conner and Folkerth et al. reported the comprehensive neuropathological examination and clinicopathological correlation of the first large case series of brains from victims of IPV.4
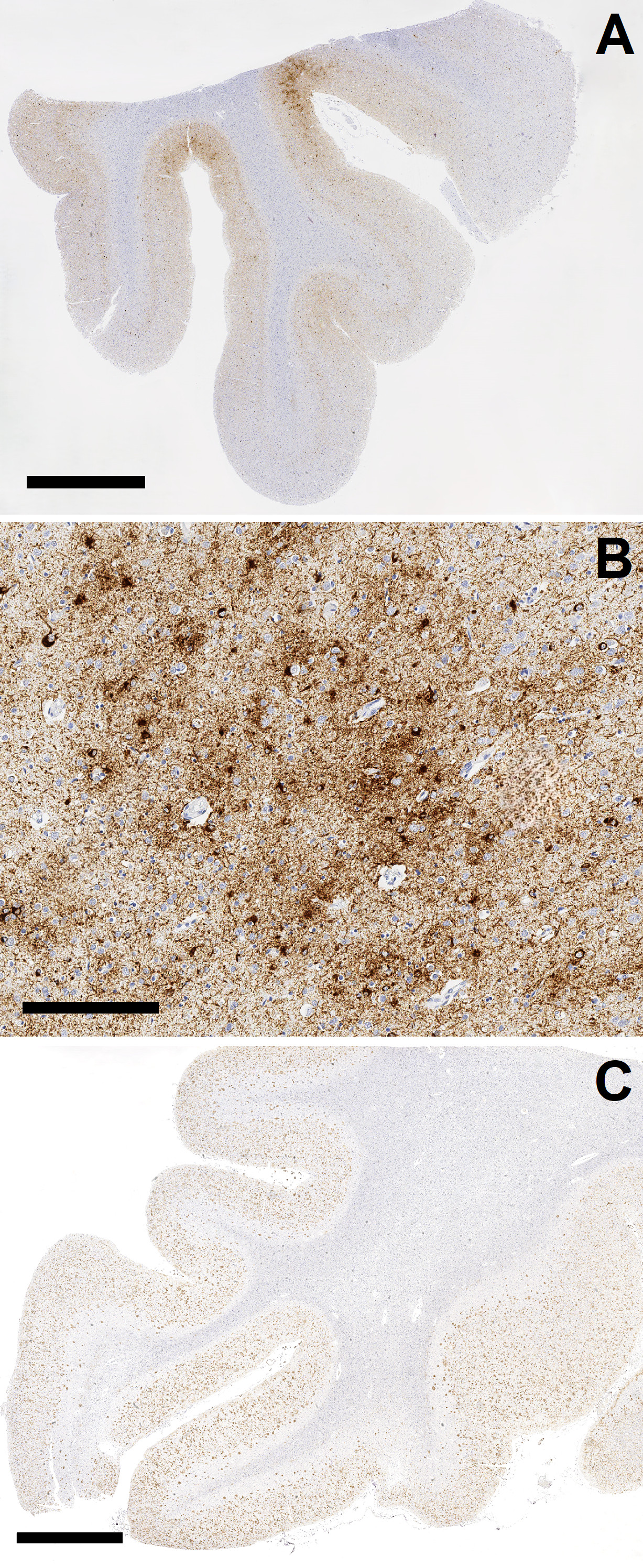
Figure 1. Total PubMed listings regarding intimate partner violence (IPV) and related terms, by year. The authors used a multi-prong approach to address this question. First, they prospectively identified a total of 14 brains from documented IPV cases which had come to the New York City Office of Chief Medical Examiner over a 24-month period. These cases were comprehensively examined neuropathologically, and all available medical records, medicolegal records, and data available from post-mortem interviews from next-of-kin were reviewed. The fourteen prospectively gathered cases were supplemented by the retrospective review of neuropathological material from 70 additional cases derived from additional victims of IPV. The fourteen prospectively gathered cases, as could be expected, had complex histories associated with IPV, including known TBI in six cases, non-fatal strangulation in four, epilepsy in seven, and some history of cerebrovascular, neurological, or psychiatric condition (related to IPV or otherwise) in thirteen. In this group, evidence of prior TBI, such as old contusions or subdural membranes, was identified in eight cases, and evidence of more recent injuries was identified in six. Amyloid Precursor Protein (APP) immunohistochemistry demonstrated evidence of axonal injury in regions susceptible to traumatic diffuse axonal injury in nine cases; further, these regions were highlighted by perivascular or parenchymal microgliosis (CD68 immunohistochemistry) and/or iron deposition (Perls stain) in twelve cases, many of which were in the absence of hypertensive arteriolosclerotic disease. This study illustrates that the complexity of IPV translates to the variable development of different corresponding neuropathologies, most of which are related to direct anatomic disruptions from trauma such as hemorrhage, contusion, and axonal injury. Importantly, and despite high rates of neuropsychiatric symptomatology in the cohort, comprehensive examination did not reveal evidence of CTE in any of the 14 cases. This finding was corroborated in the second portion of the study analyzing 70 archival cases of IPV, which similarly did not reveal a single case of CTE. Therefore, this study also suggests that TBI sustained in the context of IPV, similar to that sustained in other non-athletic contexts wherein impact TBI occurs but is less predictable and certainly less repetitive,5 is often of an insufficient dose and/or mechanism to produce CTE neuropathology. Moreover, this data further suggests that CTE does not provide a “catch-all” or common denominator accounting for high proportions of chronic neuropsychiatric sequalae following TBI. As such, we also feel that this study serves as an important opportunity to urge caution regarding the overinterpretation of isolated cases reports in our research practices, particularly as it concerns diseases with high public interest such as CTE. CTE in young contact sports athletes The large majority of CTE cases have been characterized in older age groups and/or in athletes with long careers, particularly those with an elite level of sport participation.6 However, a lingering question concerns development of CTE at young ages and the risk of developing CTE from lower level athletic exposures (e.g., high school sports). Among the most impactful studies with respect to public awareness in 2023 was written by Dr. Ann Mckee and colleagues, titled “Neuropathologic and Clinical Findings in Young Contact Sport Athletes Exposed to Repetitive Head Impacts”.7 The study describes the neuropathologic evaluation for CTE, with use of the “McKee” staging system to assess severity,8 in a convenience sample of 152 donated brains from former contact sports athletes and who were under the age of 30 years at death (range = 13–29 years). The majority of the series (60.5 %) had a history of participation in American football, with the average duration of football play being 10.29 years, and the average age at which football participation began being 9.25 years. Of the 152 cases, 128 were classified as “amateur athletes” that did not play at a semiprofessional or professional level but rather played in youth, high school (most frequent), or up to collegiate levels. The authors report that CTE was present in 63 of 152 cases (41.4 %), the large majority (95.2 %) of which had minimal/mild pathology (McKee stages I or II). 45 of the 63 CTE cases were from amateur athletes. Consistent with what has been observed and reported in the literature,6 the brains with CTE were from individuals who were significantly older, and had significantly longer durations of play than those without CTE. However, the youngest individual reportedly diagnosed with CTE in this study was 17 years old. The authors of the study have made an important contribution with the largest series of athletes in this young age group to be assessed for CTE pathology, and identifying pathologic features at a relatively high frequency. However, apart from the largely unavoidable limitations of ascertainment bias in a convenience sample derived from a brain bank dedicated to TBI research, there are a number of concerns with this study that we care to highlight. First, though McKee et al state that evaluation for CTE in this study was done in accordance with the most recent National Institute of Neurological Disorders and Stroke/National Institute of Biomedical Imaging and Bioengineering (NINDS/NIBIB) consensus criteria,9 there is no detail provided with respect to the extent or manner of sampling that was performed in these cases. In addition, the authors provide their interpretation of the pathognomonic lesion of CTE as occurring “typically”, and “most frequently” at the depth of a cortical sulcus, which stands in notable contrast to the NINDS/NIBIB consensus document which states that a pathognomonic lesion of CTE must be at a sulcal depth by definition.9 This raises concern about how these cases were being diagnosed, as an especially liberal approach may very well have led to over-diagnosing CTE in these mildly affected cases. Accordingly, it will be hard to relate the data reported here to other published studies. It is also important to express concern with how the lay media has approached this study. Indeed the publication garnered nationwide media attention, which involved the publication of several anecdotal stories involving very young athletes who had died by suicide, with the implication that contact sports participation and CTE were contributory, if not causal.10-12 These media articles were written in this manner despite the fact that the study published by McKee et al identified no correlation with clinical symptoms (including neurocognitive and psychiatric symptoms) or cause of death (including suicide) with CTE when compared to cases without CTE, which itself is consistent with other recently published data.13 How media outlets may ultimately choose to interpret scientific articles is often out of our control. However, as neuropathologists who may concern ourselves with CTE or other neurologic disorders that are within the public eye, we must be especially careful with how we communicate to the media and the lay public at large when given the opportunity, so as to avoid mischaracterization and/or unnecessary hyperbolizing of our findings. Evidence of vascular injury and remodeling at sulcal depths in CTE Next, we would like to highlight the collective efforts of two separate studies, both largely focused out of the Boston University CTE center and the Veterans Administration Healthcare system in Boston, which have identified evidence of chronic vascular injury and remodeling concentrated at sulcal depths in cases of CTE, demonstrating evidence that vascular insults may be intricately tied to the disease. First, in a study that investigated the theory that microvascular and associated blood brain barrier damage at sulcal depths may be involved in the pathogenesis of CTE, authors Kirsch et al. published a large immunohistochemical and immunoassay-based study using brain tissues from cohorts which included high stage CTE, low stage CTE, and CTE-negative controls with and without history of repetitive TBI.14 Most of those with a history of repetitive TBI were exposed via contact sports. A total of 156 cases were included in the immunoassay portion of the study, which quantified markers of vascular injury and inflammation, including intercellular adhesion molecule 1 (ICAM1), vascular cellular adhesion molecule 1 (VCAM1), and C-Reactive Protein (CRP) in samples from the dorsolateral frontal region; the authors proceeded to measure for associations between these quantifications and extent of repetitive TBI exposure (years of contact sports played), evidence of microgliosis, and p-tau pathology. A smaller sampling of 57 cases with high stage CTE, low stage CTE, and no CTE were evaluated histologically via immunohistochemistry with antibodies including for ICAM1, albumin, p-tau (AT8), and microglia (Iba1, CD68). The authors found that quantified levels of ICAM1, VCAM1, and CRP were significantly increased in CTE cases versus CTE-negative cases, and generally increased with CTE severity. Further, these elevated levels were statistically associated with duration of repetitive TBI exposure, increased microglial density, and overall p-tau burden. In CTE cases, particularly in high stage cases, histologic examination revealed dramatic co-localization of ICAM1 and AT8 at sulcal depths. Whether these increases relate to repetitive impact TBI exposure, p-tau aggregation, or a combination of both remains an important question. However, all cases wherein there was a history of repetitive TBI demonstrated extravascular albumin staining in both gray and white matter, in contrast to none of the of the cases without a history of repetitive TBI, suggesting chronic compromise to vascular integrity and the blood-brain barrier following this exposure. In the second study, authors Rosen et al. imaged vascular microstructure in passively cleared (SHIELD tissue processing) and tomato lectin-stained dorsolateral frontal samples from 41 total cases, including donors with high stage CTE, low stage CTE, and no CTE (44 % of which had repetitive TBI history) using fluorescence microscopy.15 Again, the majority of the CTE cases corresponded to TBI from contact sports participation. Among the factors evaluated in different cortical regions (e.g. sulcal depth versus gyral crest) included assessments for vessel branch density (the number of vessel branches per volume of the image stack), and vessel fraction volume (the volume of blood vessels per volume of the image stack). First, CTE cases demonstrated evidence of vascular remodeling characterized by significantly increased vascular branch density at the sulcal depths compared to non-CTE controls. Further, there were statistically significant differences in both vessel branch density and vessel fraction volume at sulcal depths versus the gyral crest in CTE cases that were not observed in controls. Finally, AT8 staining density correlated positively with vessel fraction volume at the sulcus, though this was independent of CTE status. These two studies collectively demonstrate that chronic vascular injury is an important component of repetitive, mild TBI and its predilection to sulcal depths with remarkable correlation with p-tau deposition in CTE offers a tantalizing theory for CTE pathogenesis. Indeed, vascular/blood-brain-barrier injury from TBI could induce insufficient delivery of nutrients (including oxygen) and removal of waste products, and promote a malnourished/hypoxic, pro-inflammatory, and even neurodegenerative environment in which p-tau can accumulate. Further, while authors Rosen et al. postulate that the changes in vascular branching and volume at sulcal depths in CTE may be accounted for by atrophy in the sulcus relative to the gyral crest, it has also been demonstrated that chronic vascular insufficiency triggers pro-angiogenic proteins, such as vascular endothelial growth factor.16,17 Nonetheless, it cannot be stated with certainty whether the processes of vascular injury and remodeling are pathogenically linked to CTE rather than being co-occurring/parallel processes in locations susceptible to impact TBI.18 More research is needed. Assessing multiple concurrent tauopathies in the setting of CTE: a way forward? The second, and still the most recent, NINDS/NIBIB consensus for the neuropathological diagnosis of CTE made important strides regarding diagnostic approach, in that it refined the definition of the pathognomonic lesion for CTE.9 Specifically, the newest consensus definition of CTE defines it as a disease of perivascular neuronal tau deposition at sulcal depths, with or without glial tau; the definition was adjusted in part to allow for a more careful delineation of CTE from age-related tau astrogliopathy (ARTAG), which as the name suggests is limited to tau accumulation in astrocytes but also tends to be limited to superficial cortex when it involves sulcal depths. Beyond the discernment of ARTAG, the consensus paper stated that the interpretation of CTE in the context of other multiple, concurrent neurodegenerative diseases is a matter of future consideration, and specifically made “no assertion” in this regard. However, especially in brains from older individuals with CTE, co-pathologies which include other tauopathies (e.g. Alzheimer disease [AD], primary age-related tauopathy, progressive supranuclear palsy [PSP], etc.) are common,8,19 and this creates a dilemma in neuropathology practice. Though multiple co-occurring tauopathies can certainly be diagnosed (Figure 2), given the degree of overlap between CTE distribution and that of other tauopathies, distinguishing (i.e. determining which p-tau aggregates belong to which disease) and thus staging any individual tauopathy in this circumstance under current guidance can be extraordinarily difficult, if not impossible. Importantly, this issue also precludes many aspects regarding the clinicopathological correlation of CTE and the clinical validation of pathological staging criteria.
Figure 2. Chronic traumatic encephalopathy (CTE) overlap with Alzheimer disease. While we hope for a third NINDS/NIBIB Consensus effort in the near future that could begin to address this unresolved issue, in 2023 authors Sorrentino et al. investigated a solution and wrote a brief manuscript detailing a study in which they used an immunohistochemical panel of multiple antibodies for p-tau on diagnostic material from a small series of cases of AD, CTE, PSP, and controls.20 Antibodies utilized in the panel included AT8 (p-tau at S202 and T205), AT180 (p-tau at T231), PHF1 (p-tau at S396 and S404), and MC1, which is an antibody specifically raised against p-tau in AD.21 The authors found little discernable difference in cases of AD, CTE, and PSP labeled with AT8, AT108, and PHF1. However, interestingly, MC1 immunostaining was minimal to absent in cases of CTE and PSP in comparison to the strong staining identified in the AD case as rated subjectively and also calculated by assessment of p-tau inclusions per three 10x microscopic fields (p = ≤ 0.0002). In this study, the authors demonstrate evidence that the conformation of p-tau in AD is sufficiently different so as to potentially allow the differentiation of AD from other tauopathies, importantly CTE, through the use of MC1 immunohistochemistry. This study, though small, therefore provides promise for a simple and practical means to potentially address major diagnostic issues concerning CTE when concurrent with multiple tauopathies. More investigation with mixed cases is needed, particularly given that, at the time of this writing, the discussion regarding mixed neurodegenerative pathologies in brains from individuals exposed to repetitive TBI continues to grow.22 The relationship between TBI, CTE, and TDP43 pathology TDP43 pathology has been described in a relatively small number of mostly advanced CTE cases, including descriptions of neocortical deposits (sometimes at sulcal depths, coincident with CTE pathognomonic lesions) as well as deposits in limbic structures, sometimes coincident with hippocampal sclerosis (HS).23-27 As it concerns CTE with concurrent limbic TDP43 pathology and HS, the precise relationship between these entities has been uncertain, as has been how limbic TDP43 pathology and HS may differ in the context of CTE versus that of aging alone (i.e., the impact of TBI on these lesions). In 2023, authors Nicks et al. published a large cohort study to begin to address this gap in knowledge.28 The authors began with a 401-case cohort of cases with CTE and known TBI history, predominantly from contact sports participation (mostly American football). The mean age of the cohort was 62.2 years, with a range of 20–100 years. The authors found evidence of TDP43 pathology in 43.3 % of the CTE cases, and HS in 23.4 % of cases. Regardless of whether or not HS was identified, TDP43 pathology was most common in the limbic regions (93 % of TDP43-positive cases), and less so in the examined frontal cortex (47 % of TDP43-positive cases, including 7 % which only had TDP43 accumulations in the frontal cortex in the absence of limbic pathology). Perhaps expectedly, the most numerically significant factor between CTE cases with and without HS was age; the average age in the CTE group with HS was 77 years (range 52–100), and without HS was 57.7 (range: 20–91). Interestingly, however, another statistically significant factor that emerged in the study was duration of contact sport participation, which was significantly longer in the CTE group with HS as opposed to without (p=0.029). As a means of comparison, the group also assessed differences between the CTE cohort and a 33-donor cohort diagnosed with HS related to aging, without a history of TBI and without CTE (average age = 86.6 years). Among the notable observations was that the CTE group with HS had a substantially greater proportion of cases with TDP43 inclusions within both limbic and frontal regions (41.5 %) in comparison to the group with HS but no CTE (18.2 %), which was particularly interesting given that the CTE group had an average age of death that was 10 years younger than the HS group without CTE. Also interesting was that the distribution of TDP43 pathology in the hippocampus differed between the groups. In the CTE group with HS, the CA1 region and dentate gyrus were statistically less likely to be involved by any TDP43 pathology, and the CA2 region was more severely involved TDP43 pathology. With this study, the authors demonstrate a high rate of TDP43 pathology in aged individuals with CTE, and provide evidence that CTE, and/or repetitive TBI, may exacerbate or accelerate age-related TDP43 pathology with or without HS, and/or may be a risk factor for the development of independent, and perhaps different, TDP43 pathology. The latter notion becomes particularly interesting when considering another 2023 article which identified TDP43 inclusions in the inner nuclear layer of 6/8 retinas from cases with CTE, and in only 1/8 of the age-matched controls.29 We are excited to see what the future holds for these new developments. Repetitive TBI elicits pathology in C9orf72 transgenic mice In our continued efforts to understand the link between amyotrophic lateral sclerosis (ALS) and frontotemporal dementia, the identification of C9orf72 hexanucleotide gene expansion as the most common genetic association between these disorders was a landmark discovery.30,31 Interestingly, links between repetitive TBI and ALS have also been suggested, and bolstered by evidence epidemiologically, which itself includes demonstrations that a history of multiple TBIs is associated with a three-fold increased risk of ALS, and that incidence and mortality from ALS is nearly four times higher in professional American football players in comparison with the general population.32-34 Questions surrounding how TBI may impact development of pathology in the setting of a known genetic risk for frontotemporal dementia/ALS should therefore be explored. In a 2023 article published in Brain, authors Kahriman et al. investigated the effect of repeated, mild TBI on producing pathology in a transgenic hemizygous and homozygous C9orf72 expansion mouse model.35 The authors experimented on 63 total mice, including transgenic and non-transgenic lines, 34 of which were subjected to repetitive, mild, closed-head TBI using a weight-drop impact model and 29 of which were sham. The TBI model delivered over 5 consecutive days, and the brains were removed for neuropathologic examination 52 weeks following exposure. In the interim period, mice were subjected to a variety of neurologic and behavioral testing. Alterations in neurologic and behavioral testing during the long survival period were observed in the transgenic, TBI-subjected mice, which included but were not limited to significantly reduced grip strength, and behaviors that were similar to some phenotypes of human ALS. Neuropathologic findings in these mice were also particularly striking. Histological examination included chromogenic immunohistochemistry and immunofluorescent staining for antibodies including TDP43, glial fibrillary acidic protein (GFAP), Iba1 and CD68, neurofilament (SMI-312), synaptophysin (a pre-synaptic marker) and PSD-95 (a post-synaptic marker), and neuronal markers NeuN, Tuj1 (Beta-III tubulin), and MAP2. Examination revealed that brains from the TBI-exposed, transgenic mice suffered significantly more neuronal loss in the cortex along with axonal and synaptic marker loss and evidence of widespread microglial activation in comparison with controls. These findings were seen coincidently with dramatic cytoplasmic mislocalization of TDP43 in the remaining neurons of the cortex, which was not observed in either the non-transgenic mice which sustained TBI or the transgenic sham mice following the 52-week survival period. With these findings, the authors demonstrate evidence that repetitive TBI is sufficient to initiate, exacerbate, and accelerate the development of frontotemporal dementia/ALS-like pathology in the setting of genetic predisposition with C9orf72 expansion in a mouse model. The findings are most informative with regard the relationship of TBI and ALS, and also have implications with regard to development and implementation of therapy for TDP43 pathology and/or TBI. Patterns of head and neck injury in fatal underbody blast exposures TBI is considered a “signature wound” of modern warfare, and blast exposure is acknowledged as the most common source of TBI in the military context.36,37 Among the more common causes of military blast-related morbidity and mortality is so-called underbody, or “mounted” blast exposure. Underbody blast exposure, which became notable due to the increased use of improvised explosive devices in the most recent wars in the Middle East, is sustained when an explosive is detonated under a vehicle. This causes the vehicle, its contents/occupants to be propelled upwards in a rapid acceleration event that further includes impacts between vehicle occupants and other occupants, vehicle contents, and/or the inner walls of the vehicle. The precise mechanism(s) by which fatal central nervous system injuries occur in this context, and/or constellations that these injuries tend to occur in, have been poorly understood. Ashworth et al. studied post-mortem computed tomography scans of 46 underbody blast fatalities which occurred from 2007–2013 among British military personnel.38 The authors identified that the most common pathology identified within or of the cranium was subarachnoid hemorrhage (80.4 % of total cases), followed by skull fracture (63 %, including 10.9 % of cases with an “eggshell” pattern of skull fracture reflecting direct head impact), 4th ventricular hemorrhage (63 %), and contusions (28.3 %). Other injuries observed less frequently included subdural hematoma, epidural hematoma, brainstem hemorrhage, and pneumocephalus. Radiologic evidence of diffuse axonal injury was uncommon (1 case). Examination of the spine frequently revealed fractures, being most common in the lower spine (lumbar fractures in 52.2 % of cases, sacral in 50 %), followed by the thoracic and cervical. In examining the constellation of injuries to the central nervous system and its surroundings, together with secondary injuries to other regions (e.g. thorax, abdomen, limbs), the authors hypothesized five categories/patterns of potential head and neck injury in fatal underbody blasts. Briefly, these constellations are published as follows:
The information in this study may be particularly important to medical examiners and/or neuropathologists who practice in a forensic setting. Further, the article may also serve to emphasize the growing utility of post-mortem imaging in diagnostic practice. Beyond the scope of neuropathology, understanding mechanisms by which the central nervous system can be injured in various settings of blast exposure is critical for diagnosis and care, including triage efforts, and also for the development of protective measures and mitigation. Chronically altered brain metal homeostasis following repetitive impact TBI Alterations in the content and metabolism of metals including iron, zinc, and copper have been identified in the context of aging and Alzheimer disease, as well as Parkinson’s disease, and have been implicated with misfolding of pathogenic proteins in neurodegeneration.39-43 While a small number of studies have reported iron accumulation following repeated traumatic brain injury,44-46 there has been little investigation with regard to alterations in iron regulatory proteins and the potential association of metal dysregulation with neurodegeneration in the context of TBI. Paul Adlard’s laboratory had previously shown altered brain metals following a single impact TBI, and demonstrated increased iron levels in the subacute period following repeated impact TBI, in mice.47,48 As a follow-up, they investigated chronic alterations in metal content, iron regulatory proteins, and neurodegenerative proteins in a mouse model of mild, repetitive impact TBI with prolonged survival within a study they published in 2023.49 The authors subjected groups of three-month-old mice to either single or repeated (five instances, each 48 hours apart) mild, closed-head impact TBI, and compared to corresponding sham controls. In order to demonstrate evidence of ongoing genetic changes following TBI, RNA sequencing and differential gene expression analysis was performed on ipsilateral and contralateral cerebral hemispheres from mice sacrificed following 6 months of survival. In the mice with 12 months of survival, ipsilateral and contralateral cortical samples were subjected to metal analysis using inductively coupled plasma-mass spectrometry (ICP-MS) and Western blot analysis using a list of antibodies related to iron regulatory proteins, to tau and tau regulatory proteins, and to amyloid precursor protein. The authors identified significant alterations in metal content and regulatory proteins in the mouse brains exposed to repetitive TBI. First, they found increases in ferritin and divalent metal transporter 1 (DMT1, a cytosolic iron transport protein), and a decrease in transferrin receptor in the ipsilateral cortex. This signature would typically imply iron excess; however, these findings were observed in the context of a downward trend in total iron content, suggesting perhaps that iron had previously been elevated but then restored with a regulatory response. Conversely, in the contralateral cortex of mice exposed to repetitive TBI, there were no significant differences in iron regulatory proteins and rather there was an increase in iron content, which could suggest iron regulatory failure on the contralateral side. This was also coincident with increases in total zinc and copper on the contralateral side, which was not observed on the ipsilateral side. It is unclear what may account for the differences between the hemispheres, but nonetheless the authors do demonstrate chronic alterations in brain metals and related metabolism following repeated TBI. Interestingly, there were no significant transcriptional or translational changes in tau or tau regulatory proteins found in either hemisphere following repeated TBI with 6- and 12- months of survival, respectively. Given the lack of this association, and the association between the measured metals and adverse neurologic sequelae/neurodegeneration, the authors posit that many of the significantly increased neurobehavioral deficits that were observed in the repeated TBI mice during the survival period were at least partially contributed to by metal excess. As such, the authors suggest that chelation therapy following repeated TBI exposure as a means to potentially prevent long-term adverse outcomes is worthy of further investigation. Blast exposure damages the neurovascular unit in mice Between the years 2000 and 2023, there were almost 500,000 individuals with new diagnoses of TBI among United States military personnel recorded by the Defense Health Agency of the United States Department of Defense, the overwhelming majority (82.2 %) of which were classified as mild TBI.50 One aspect of the military experience that is particularly unique in comparison to the civilian population is exposure to repetitive blast overpressures in the context of both live combat and in training circumstances. Of increasing recognition is the potential of so-called “low-level blast exposure” (LLB), i.e. that which may be generated by firing heavy weapons, breaching exercises, or other low-intensity explosives, to produce mild TBI or subclinical damage to the brain with potential chronic implications.51-53 However, our understanding of the pathophysiology of such injury is still in development. The neurovascular unit is a functional unit of the brain that is critical in the management of cerebral blood flow and maintenance of the blood-brain barrier, being composed of the delicate and complex interplay of endothelial cells and their tight junctions, perivascular astrocytes (particularly astrocytic endfeet), pericytes, basement membranes, and finally adjacent microglia and neurons. Certainly, insults to the neurovascular unit are implicated in a wide variety of neurological disease states.54 In 2023, authors Li et al. tested a hypothesis that LLB could induce ultrastructural abnormalities of the neurovascular unit using a mouse model.55 The authors exposed a group of anesthetized mice to a single open-field LLB, during which no head or body movements were observed. Among the several studies performed on the brain tissues following sacrifice, examination of ultrastructure by transmission electron microscopy (EM) in mice sacrificed 7 and 30 days following LLB exposure produced many of the most interesting findings, revealing a myriad of changes which suggest injury to multiple constituents of the neurovascular unit. In summary, the statistically significant findings in comparison to controls included drop-out of pericyte surface area and associated coverage of endothelial cells at 30 days survival, endothelial swelling at 7 days survival with loss and/or discontinuity of tight junctions at both 7 and 30 days survival, thickening of basement membranes at 7 and 30 days survival, swelling of astrocytic endfeet at 7 and 30 days survival, and finally detachments of astrocytic endfeet with one another as well as with basement membranes. The above changes were coincident with marked luminal constriction relative to controls at 7 days survival, and dilation at 30 days survival, potentially indicating the presence and reversal of vasospasm. Many unquantified morphological changes were also observed in comparison to controls, including vacuolization of numerous neurovascular unit constituents (pericytes, endothelial cells, basement membranes, astrocytic endfeet) potentially indicative of cellular edema, detachments between endothelial cells and bulging of endothelial cytoplasm into the lumen, and bulging and fragmentation of basement membranes. With these findings, the authors demonstrate evidence of significant neurovascular unit damage and loss of structural integrity of the blood-brain barrier in the short and intermediate survival period that follows just a single LLB exposure in mice. This, of course, may have profound implications with regard to military Service Members who may experience many multitudes of these exposures, and at short intervals, throughout the length of a career. Certainly, studies with long-term survival and neurobehavioral analysis are necessary to determine the potential chronicity of these changes, or to determine how these changes may evolve over time. Human autopsy studies with this research question are also paramount. Nonetheless, this study serves as an indication that blast overpressure exposure in the military context, no matter how low the intensity, should be monitored and minimized where possible. Muhammad Ali had young-onset, idiopathic Parkinson disease On June 3rd, 2016 Muhammad Ali died of sepsis. Famously, Ali denied autopsy owing to his Muslim faith. During approximately 34 years preceding his death, Ali suffered from a progressive parkinsonian movement disorder that was and has remained among the most widely discussed anecdotes in neurodegeneration. Particularly accelerated by the resurgence of dementia pugilistica as CTE, speculation regarding the role of Ali’s illustrious boxing career on his manifestation of parkinsonism has escalated. Indeed, many lay media depictions have, at least, made implications of a strong role of TBI,56,57 with one lay book that even speculates CTE.58 Continuing with what has become a theme in this writing, we provide the readership one last precautionary message regarding overinterpretation and undue speculation on individual cases and bring to awareness an article published in 2023 that provides for us a succinct, yet comprehensive medical account of Ali’s clinical course. This leads to an important conclusion: in the absence of an autopsy, all of the information available to us indicates that Muhammad Ali suffered from classic, early-onset, idiopathic Parkinson disease.59 Neurologists Michael Okun, Helen Mayberg, and Mahlon DeLong, all of whom were involved in Muhammad Ali’s clinical care at Emory University, give us an account of the two decades of clinical follow-up with Ali at the institution to support their conclusion. In brief, below is a bulleted summary:
Though the natural history of Ali’s disease is quite characteristic for idiopathic Parkinson disease, several additional diagnostic findings provide further support. First, fluorodeoxyglucose positron emission tomography (PET) and fluorodopa F18 PET scans performed in 1997 and 1998 demonstrated bilateral striatal activity and low striatal uptake, respectively, both of which are consistent with and classic for Parkinson disease in contrast to post-traumatic parkinsonism. Polysomnography to investigate Ali’s sleep dysfunction revealed rapid-eye movement sleep behavioral disorder which, though not specific, is most classically associated with synucleinopathies including Parkinson disease. Finally, the clinical feature that draws the most convincing contrast between Parkinson disease and post-traumatic parkinsonism or CTE is the fact that Ali’s symptoms were “clearly” and “substantially” responsive to levodopa. In summary, Muhammad Ali experienced a 34-year chronic, progressive course that followed a classic natural history of Parkinson disease, with compatible (if not classical) imaging, and that was responsive to levodopa. This is unlike posttraumatic parkinsonism which can be transitory, often shows kinetic tremor, and is not characteristically responsive to levodopa. Though in the absence of postmortem examination it can never be stated with certainty if Ali did, in fact, have CTE neuropathological changes, the available (and rather complete) clinical evidence strongly suggests that at least his symptomatology was driven by early-onset, idiopathic Parkinson disease. In Memoriam: Dr. Byron A. Kakulas We would like to end this years’ listing to honor the legacy of Dr. Byron A. Kakulas. In January, 2023, Dr. Kakulas (Emeritus Professor, University of Western Australia) died at age 90 in his native city, Perth, Western Australia. While he was mostly known for his work on muscular dystrophy, he was also a world authority on the neuropathology of spinal cord injury. His interest in spinal cord injury began early in his career in neuropathology and over the years he accumulated and studied a remarkably large number of anatomic specimens with this lesion. For those with interest in spinal cord injury, Sir Ludwig Guttmann is considered to be a towering figure in the history of its treatment. Guttmann was an early pioneer in changing the clinical outcome for spinal cord injury patients and, among other things, introduced sports participation in their rehabilitation process. Perhaps the foremost acknowledgment of one’s standing in the field of spinal cord research is to be asked to give the Ludwig Guttmann Memorial Lecture at the annual meeting of the International Spinal Cord Society. In September, 2004, Byron Kakulas gave the Guttmann Lecture entitled: “Neuropathology: the foundation for new treatments in spinal cord injury”.61 We believe he is the only neuropathologist to be so honored. His lecture has been published and remains a remarkable document, summarizing his findings and thoughts on the subject based on examining a total of 588 spinal cord injury cases! For those involved in studies of spinal cord injury who have not read this paper (and his others on the subject), it is highly recommended and shows Professor Kakulas’ immense talents as a neuropathologist, and his depth of knowledge and understanding of the subject. It is truly a classic paper and as he clearly demonstrates: “In the context of finding a cure for spinal cord injury, the first and foremost requirement is an in-depth knowledge of the disorder in neuropathological terms with the complexity of the spinal cord appreciated.” To read further on Professor Kakulas’ life and career in neuropathology, the reader is referred to his obituary published in Free Neuropathology (see https://doi.org/10.17879/freeneuropathology-2023-4820).62 Byron Kakulas was a superb neuropathologist, a very creative scientist, a charismatic mentor to many, and a man who was devoted to his Greek heritage and to his loving extended family. He was also a cherished friend of one of the authors (DPP, Figure 3). He will be greatly missed.
Figure 3. In memory of Dr. Byron Kakulas Conflicts of Interest Statement The authors do not have any conflict of interest to declare. Funding Statement The work of the authors is supported by the DoD/USU Brain Tissue Repository and Neuropathology Program, USU award #HU00012120007 (HJF award# 312159-1.00-66531). Disclaimer The information/content, conclusions, and/or opinions expressed herein do not necessarily represent the official position or policy of, nor should any official endorsement be inferred on the part of, Uniformed Services University, The Department of Defense, United States Uniformed Services, or the United States Government. References 1. Sardinha L, Maheu-Giroux M, Stöckl H, Meyer SR, García-Moreno C. Global, regional, and national prevalence estimates of physical or sexual, or both, intimate partner violence against women in 2018. Lancet. 2022 Feb 26;399(10327):803-813. 2. Roberts GW, Whitwell HL, Acland PR, Bruton CJ. Dementia in a punch-drunk wife. Lancet. 1990 Apr 14;335(8694):918-9. 3. Danielsen T, Hauch C, Kelly L, White CL. Chronic Traumatic Encephalopathy (CTE)-Type Neuropathology in a Young Victim of Domestic Abuse. J Neuropathol Exp Neurol. 2021 Jun 4;80(6):624-627. 4. Dams-O'Connor K, Seifert AC, Crary JF, Delman BN, Del Bigio MR, Kovacs GG, Lee EB, Nolan AL, Pruyser A, Selmanovic E, Stewart W, Woodoff-Leith E, Folkerth RD. The neuropathology of intimate partner violence. Acta Neuropathol. 2023 Dec;146(6):803-815. 5. Priemer DS, Iacono D, Rhodes CH, Olsen CH, Perl DP. Chronic Traumatic Encephalopathy in the Brains of Military Personnel. N Engl J Med. 2022;386(23):2169-2177. 6. McKee AC, Stein TD, Huber BR, Crary JF, Bieniek K, Dickson D, Alvarez VE, Cherry JD, Farrell K, Butler M, Uretsky M, Abdolmohammadi B, Alosco ML, Tripodis Y, Mez J, Daneshvar DH. Chronic traumatic encephalopathy (CTE): criteria for neuropathological diagnosis and relationship to repetitive head impacts. Acta Neuropathol. 2023 Apr;145(4):371-394. 7. McKee AC, Mez J, Abdolmohammadi B, Butler M, Huber BR, Uretsky M, Babcock K, Cherry JD, Alvarez VE, Martin B, Tripodis Y, Palmisano JN, Cormier KA, Kubilus CA, Nicks R, Kirsch D, Mahar I, McHale L, Nowinski C, Cantu RC, Stern RA, Daneshvar D, Goldstein LE, Katz DI, Kowall NW, Dwyer B, Stein TD, Alosco ML. Neuropathologic and Clinical Findings in Young Contact Sport Athletes Exposed to Repetitive Head Impacts. JAMA Neurol. 2023 Oct 1;80(10):1037-1050. 8. McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, Lee HS, Wojtowicz SM, Hall G, Baugh CM, Riley DO, Kubilus CA, Cormier KA, Jacobs MA, Martin BR, Abraham CR, Ikezu T, Reichard RR, Wolozin BL, Budson AE, Goldstein LE, Kowall NW, Cantu RC. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013 Jan;136(Pt 1):43-64. 9. Bieniek KF, Cairns NJ, Crary JF, Dickson DW, Folkerth RD, Keene CD, Litvan I, Perl DP, Stein TD, Vonsattel JP, Stewart W, Dams-O'Connor K, Gordon WA, Tripodis Y, Alvarez VE, Mez J, Alosco ML, McKee AC; TBI/CTE Research Group. The Second NINDS/NIBIB Consensus Meeting to Define Neuropathological Criteria for the Diagnosis of Chronic Traumatic Encephalopathy. J Neuropathol Exp Neurol. 2021 Feb 22;80(3):210-219. 10. Musa A. “A living hell inside of my head”: For first time, more advanced stage of CTE diagnosed in teen football player. CNN. 2023 Nov 21. 11. Pohlman D. Startling new evidence reveals deadly brain disease strikes young athletes at alarming rates. CW Columbus. 2023 Dec 14. 12. Bracken K, Branch J, Laffin B, Lieberman R, Ward J. They started playing football as young as 6. They died in their teens and twenties with CTE. The New York Times. 2023 Nov 17. 13. Iverson GL, Gaudet CE, Karr JE. Examining Suicidality in Adolescents Who Have Sustained Concussions. J Neurotrauma. 2023 Apr;40(7-8):730-741. 14. Kirsch D, Shah A, Dixon E, Kelley H, Cherry JD, Xia W, Daley S, Aytan N, Cormier K, Kubilus C, Mathias R, Alvarez VE, Huber BR, McKee AC, Stein TD. Vascular injury is associated with repetitive head impacts and tau pathology in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. 2023 Jan 20;82(2):127-139. 15. Rosen G, Kirsch D, Horowitz S, Cherry JD, Nicks R, Kelley H, Uretsky M, Dell'Aquila K, Mathias R, Cormier KA, Kubilus CA, Mez J, Tripodis Y, Stein TD, Alvarez VE, Alosco ML, McKee AC, Huber BR. Three dimensional evaluation of cerebrovascular density and branching in chronic traumatic encephalopathy. Acta Neuropathol Commun. 2023 Jul 25;11(1):123. 16. Holmes DI, Zachary I. The vascular endothelial growth factor (VEGF) family: Angiogenic factors in health and disease. Genome Biol 2005;6:209. 17. Snyder B, Shell B, Cunningham JT, et al. Chronic intermittent hypoxia induces oxidative stress and inflammation in brain regions associated with early-stage neurodegeneration. Physiol Rep 2017;5:e13258. 18. Kerwin J, Yücesoy A, Vidhate S, et al. Sulcal Cavitation in Linear Head Acceleration: Possible Correlation With Chronic Traumatic Encephalopathy. Front Neurol. 2022 Feb 28;13:832370. 19. Lee EB, Kinch K, Johnson VE, Trojanowski JQ, Smith DH, Stewart W. Chronic traumatic encephalopathy is a common co-morbidity, but less frequent primary dementia in former soccer and rugby players. Acta Neuropathol. 2019 Sep;138(3):389-399. 20. Sorrentino ZA, Paterno G, Giasson BI, Bailes JE, Lee JM, Lucke-Wold B. Differentiating pathologic tau in chronic traumatic encephalopathy (CTE) from other tauopathies: A potential antibody panel assessment. J Neuropathol Exp Neurol. 2023 Sep 20;82(10):876-879. 21. Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997 Apr 15;48(2):128-32. 22. Saltiel N, Tripodis Y, Menzin T, Olaniyan A, Baucom Z, Yhang E, Palmisano JN, Martin B, Uretsky M, Nair E, Abdolmohammadi B, Shah A, Nicks R, Nowinski C, Cantu RC, Daneshvar DH, Dwyer B, Katz DI, Stern RA, Alvarez VE, Huber B, Boyle PA, Schneider JA, Mez J, McKee AC, Alosco ML, Stein TD. Relative Contributions of Mixed Pathologies to Cognitive and Functional Symptoms in Brain Donors Exposed to Repetitive Head Impacts. Ann Neurol. 2024 Feb;95(2):314-324. 23. Hales C, Neill S, Gearing M, Cooper D, Glass J, Lah J. Late-stage CTE pathology in a retired soccer player with dementia. Neurology. 2014 Dec 9;83(24):2307-9. 24. King A, Sweeney F, Bodi I, Troakes C, Maekawa S, Al-Sarraj S. Abnormal TDP-43 expression is identified in the neocortex in cases of dementia pugilistica, but is mainly confined to the limbic system when identified in high and moderate stages of Alzheimer's disease. Neuropathology. 2010 Aug;30(4):408-19. 25. McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW, Perl DP, Hedley-Whyte ET, Price B, Sullivan C, Morin P, Lee HS, Kubilus CA, Daneshvar DH, Wulff M, Budson AE. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. 2010 Sep;69(9):918-29. 26. Saing T, Dick M, Nelson PT, Kim RC, Cribbs DH, Head E. Frontal cortex neuropathology in dementia pugilistica. J Neurotrauma. 2012 Apr 10;29(6):1054-70. 27. van Amerongen S, Kamps S, Kaijser KKM, Pijnenburg YAL, Scheltens P, Teunissen CE, Barkhof F, Ossenkoppele R, Rozemuller AJM, Stern RA, Hoozemans JJM, Vijverberg EGB. Severe CTE and TDP-43 pathology in a former professional soccer player with dementia: a clinicopathological case report and review of the literature. Acta Neuropathol Commun. 2023 May 10;11(1):77. 28. Nicks R, Clement NF, Alvarez VE, Tripodis Y, Baucom ZH, Huber BR, Mez J, Alosco ML, Aytan N, Cherry JD, Cormier KA, Kubilius C, Mathias R, Svirsky SE, Pothast MJ, Hildebrandt AM, Chung J, Han X, Crary JF, McKee AC, Frosch MP, Stein TD. Repetitive head impacts and chronic traumatic encephalopathy are associated with TDP-43 inclusions and hippocampal sclerosis. Acta Neuropathol. 2023 Apr;145(4):395-408. 29. Phansalkar R, Goodwill VS, Nirschl JJ, De Lillo C, Choi J, Spurlock E, Coughlin DG, Pizzo D, Sigurdson CJ, Hiniker A, Alvarez VE, Mckee AC, Lin JH. TDP43 pathology in chronic traumatic encephalopathy retinas. Acta Neuropathol Commun. 2023 Sep 22;11(1):152. 30. Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Hölttä-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chiò A, Restagno G, Borghero G, Sabatelli M; ITALSGEN Consortium; Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011 Oct 20;72(2):257-68. 31. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011 Oct 20;72(2):245-56. 32. Walt GS, Burris HM, Brady CB, Spencer KR, Alvarez VE, Huber BR, Guilderson L, Abdul Rauf N, Collins D, Singh T, Mathias R, Averill JG, Walker SE, Robey I, McKee AC, Kowall NW, Stein TD. Chronic Traumatic Encephalopathy Within an Amyotrophic Lateral Sclerosis Brain Bank Cohort. J Neuropathol Exp Neurol. 2018 Dec 1;77(12):1091-1100. 33. Daneshvar DH, Mez J, Alosco ML, Baucom ZH, Mahar I, Baugh CM, Valle JP, Weuve J, Paganoni S, Cantu RC, Zafonte RD, Stern RA, Stein TD, Tripodis Y, Nowinski CJ, McKee AC. Incidence of and Mortality from Amyotrophic Lateral Sclerosis in National Football League Athletes. JAMA Netw Open. 2021 Dec 1;4(12):e2138801. 34. Pupillo E, Poloni M, Bianchi E, Giussani G, Logroscino G, Zoccolella S, Chiò A, Calvo A, Corbo M, Lunetta C, Marin B, Mitchell D, Hardiman O, Rooney J, Stevic Z, Bandettini di Poggio M, Filosto M, Cotelli MS, Perini M, Riva N, Tremolizzo L, Vitelli E, Damiani D, Beghi E; EURALS Consortium. Trauma and amyotrophic lateral sclerosis: a European population-based case-control study from the EURALS consortium. Amyotroph Lateral Scler Frontotemporal Degener. 2018 Feb;19(1-2):118-125. 35. Kahriman A, Bouley J, Tuncali I, Dogan EO, Pereira M, Luu T, Bosco DA, Jaber S, Peters OM, Brown RH Jr, Henninger N. Repeated mild traumatic brain injury triggers pathology in asymptomatic C9ORF72 transgenic mice. Brain. 2023 Dec 1;146(12):5139-5152. 36. Lindquist LK, Love HC, Elbogen EB. Traumatic Brain Injury in Iraq and Afghanistan Veterans: New Results From a National Random Sample Study. J Neuropsychiatry Clin Neurosci. 2017 Summer;29(3):254-259. 37. Bhattacharjee Y. Neuroscience. Shell shock revisited: solving the puzzle of blast trauma. Science. 2008 Jan 25;319(5862):406-8. 38. Ashworth E, Baxter D, Gibb I, Wilson M, Bull AMJ. Injuries in Underbody Blast Fatalities: Identification of Five Distinct Mechanisms of Head Injury. J Neurotrauma. 2023 Jan;40(1-2):141-147. 39. Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014 Oct;13(10):1045-60. 40. Good PF, Olanow CW, Perl DP. Neuromelanin-containing neurons of the substantia nigra accumulate iron and aluminum in Parkinson's disease: a LAMMA study. Brain Res. 1992 Oct 16;593(2):343-6. 41. Massie HR, Aiello VR, Iodice AA. Changes with age in copper and superoxide dismutase levels in brains of C57BL/6J mice. Mech Ageing Dev. 1979 Apr;10(1-2):93-9. 42. Barnham KJ, Bush AI. Metals in Alzheimer's and Parkinson's diseases. Curr Opin Chem Biol. 2008 Apr;12(2):222-8. 43. Ma Q, Li Y, Du J, Liu H, Kanazawa K, Nemoto T, Nakanishi H, Zhao Y. Copper binding properties of a tau peptide associated with Alzheimer's disease studied by CD, NMR, and MALDI-TOF MS. Peptides. 2006 Apr;27(4):841-9. 44. Bouras C, Giannakopoulos P, Good PF, Hsu A, Hof PR, Perl DP. A laser microprobe mass analysis of brain aluminum and iron in dementia pugilistica: comparison with Alzheimer's disease. Eur Neurol. 1997;38(1):53-8. 45. Uryu K, Laurer H, McIntosh T, Praticò D, Martinez D, Leight S, Lee VM, Trojanowski JQ. Repetitive mild brain trauma accelerates Abeta deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. J Neurosci. 2002 Jan 15;22(2):446-54. 46. Huang L, Coats JS, Mohd-Yusof A, Yin Y, Assaad S, Muellner MJ, Kamper JE, Hartman RE, Dulcich M, Donovan VM, Oyoyo U, Obenaus A. Tissue vulnerability is increased following repetitive mild traumatic brain injury in the rat. Brain Res. 2013 Mar 7;1499:109-20. 47. Juan SMA, Daglas M, Gunn AP, Lago L, Adlard PA. Characterization of the spatial distribution of metals and profile of metalloprotein complexes in a mouse model of repetitive mild traumatic brain injury. Metallomics. 2022 Dec 8;14(12):mfac092. 48. Portbury SD, Hare DJ, Sgambelloni C, Finkelstein DI, Adlard PA. A time-course analysis of changes in cerebral metal levels following a controlled cortical impact. Metallomics. 2016 Feb;8(2):193-200. 49. Juan SMA, Daglas M, Truong PH, Mawal C, Adlard PA. Alterations in iron content, iron-regulatory proteins and behaviour without tau pathology at one year following repetitive mild traumatic brain injury. Acta Neuropathol Commun. 2023 Jul 18;11(1):118. 50. Defense Medical Surveillance System, Theater Medical Data Store provided by the Armed Forces Health Surveillance Division. Prepared by the Traumatic Brain Injury Center of Excellence, as of February 13, 2024. 51. Woodall JLA, Sak JA, Cowdrick KR, Bove Muñoz BM, McElrath JH, Trimpe GR, Mei Y, Myhre RL, Rains JK, Hutchinson CR. Repetitive Low-level Blast Exposure and Neurocognitive Effects in Army Ranger Mortarmen. Mil Med. 2023 Mar 20;188(3-4):e771-e779. 52. Belding JN, Englert R, Bonkowski J, Thomsen CJ. Occupational Risk of Low-Level Blast Exposure and TBI-Related Medical Diagnoses: A Population-Based Epidemiological Investigation (2005-2015). Int J Environ Res Public Health. 2021 Dec 8;18(24):12925. 53. Section 735 of the James M. Inhofe National Defense Authorization Act for Fiscal Year 2023: Brain Health Initiative of the Department of Defense, United States of America. 54. Lo EH, Rosenberg GA. The neurovascular unit in health and disease: introduction. Stroke. 2009 Mar;40(3 Suppl):S2-3.Epub 2008 Dec 8. 55. Li C, Chen S, Siedhoff HR, Grant D, Liu P, Balderrama A, Jackson M, Zuckerman A, Greenlief CM, Kobeissy F, Wang KW, DePalma RG, Cernak I, Cui J, Gu Z. Low-intensity open-field blast exposure effects on neurovascular unit ultrastructure in mice. Acta Neuropathol Commun. 2023 Sep 6;11(1):144. 56. Park A. The complicated link between Muhammad Ali’s death and boxing. Time. 2016 Jun 6. 57. Burns K. Muhammad Ali (Documentary series). PBS. 2021 Sep 19-22. 58. Dixon T. Damage: The Untold Story of Brain Trauma in Boxing. United Kingdom: Hamilcar Publications. 2021 May 25. pp. 1–58. 59. Okun MS, Mayberg HS, DeLong MR. Muhammad Ali and Young-Onset Idiopathic Parkinson Disease-The Missing Evidence. JAMA Neurol. 2023 Jan 1;80(1):5-6. 60. Kriegel J, Papadopoulos Z, McKee AC. Chronic Traumatic Encephalopathy: Is Latency in Symptom Onset Explained by Tau Propagation? Cold Spring Harb Perspect Med. 2018 Feb 1;8(2):a024059. 61. Kakulas BA. Neuropathology: the foundation for new treatments in spinal cord injury. Spinal Cord. 2004 Oct;42(10):549-63. 62. Harper C, Masters CL. Vale Emeritus Professor Byron A Kakulas AO, 1932 - 2023. Free Neuropathol. 2023 Jun 22;4:4-11.
Copyright: © 2024 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |