|
|
|
Free Neuropathology 5:22 (2024) |
|
Letter |
|
Striking intraneuronal neurofilament inclusions restricted to the locus coeruleus in a patient with Creutzfeldt-Jakob disease |
|
Felix Leitner1,2, Sigrid Klotz1,2, Karoline Ornig3, Serge Weis3, Ellen Gelpi1,2 |
|
|
Corresponding author: |
|
Submitted: 29 July 2024 |
|
Keywords: Neurofilament inclusions, Locus coeruleus, Neurofilament, Neuronal cytoplasmic inclusion, Creutzfeldt-Jakob disease |
|
Introduction Creutzfeldt-Jakob disease (CJD) is the most frequent human prion disease, but it is overall a very rare, rapidly progressive neurodegenerative disease.1 Physiopathologically, prion diseases have set the basis for the concept of protein misfolding as a common mechanism for the most prevalent protein-related neurodegenerative conditions. In addition, interactions between misfolded proteins have been an intense topic of basic research in the field of neurodegeneration, as has been the analysis of concomitant “proteinopathies” in neuropathology. Several cases of CJD featuring concomitant pathologies such as Alzheimer’s disease 1,2,3 or Lewy body dementia 4,5,6 have been reported over the last years. Most of the concomitant pathologies have been attributed to age rather than to a direct effect of the prion protein7,8, although this is still controversial. Here we report the neuropathological findings of a CJD patient with striking intraneuronal inclusions that are restricted to the locus coeruleus and that display a unique immunohistochemical profile not seen in other neurodegenerative diseases. We propose that these unique intraneuronal inclusions might be associated with subtle and slowly progressing neurological symptoms like rapid-eye movement behavior disorder (RBD) or reduced cognitive flexibility. Case Presentation The patient was an 84-year old female patient who developed a rapidly progressive dementia accompanied by rigidity, dysarthria and dysphagia within a few months. Brain MRI revealed a restricted-diffusion pattern of the left basal ganglia, and the EEG showed partially steeply configured signals. The cerebrospinal fluid tests revealed an increased 14-3-3 protein concentration of over 80 000 AU/ml (reference values < 10 000 AU/ml) and a positive real-time quaking-induced conversion (RTQuIC) of the prion protein (PrP). The patient fulfilled criteria for “probable” Creutzfeldt-Jakob disease (CJD)9 and died within less than 4 months. The brain weight was 1275 gram. On gross examination, there were no signs of brain atrophy. The Circulus of Willis showed only a few non-stenosing plaques in the arterial wall. There were no signs of brain swelling with tonsillar notch. The ventricular system was not enlarged. The substantia nigra and locus coeruleus appeared to be regularly pigmented. Microscopic examination revealed the spongiform neuropil change characteristic of CJD10 as well as nerve cell loss and gliosis in the deeper cortical layers of the frontal and parietal lobes as well as in the basal ganglia, thalamus, brainstem and cerebellum. Pathological prion protein deposits (anti-PrP, Bertin Bioreagent, 12F10) were immunohistochemically detected in these regions. In cortical regions, there was a predominantly deep-laminar perineuronal pattern while the basal ganglia, the cerebellum and the nuclei of the brainstem, including the locus coeruleus and the solitary tract showed a dense synaptic deposition pattern. In addition, there were some plaque-like deposits in the neighbouring white matter and also deposits along axons and perivascularly in basal ganglia and thalamus. The morphological and immunohistochemical distribution pattern was overall well compatible with the VV2 histotype according to the 2009 Parchi classification of CJD (Figure 1).11
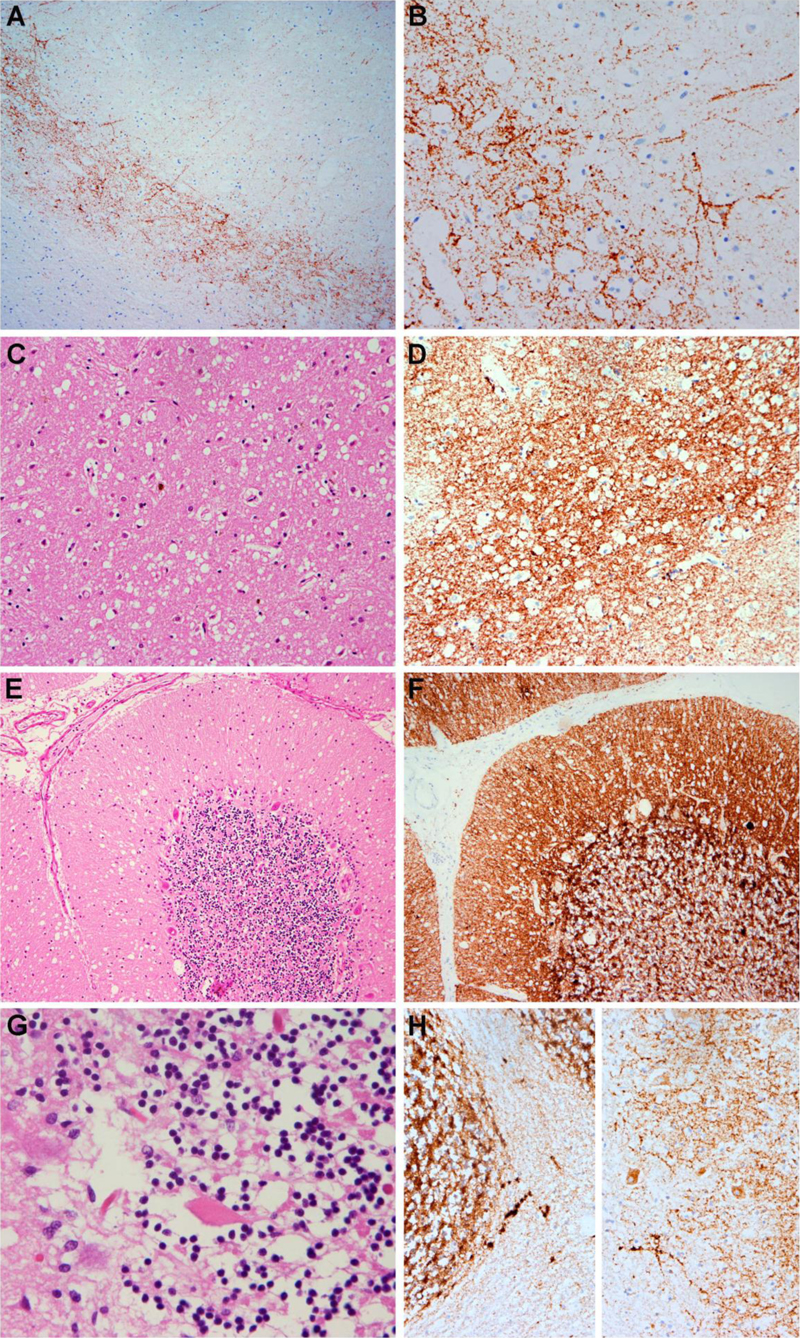
Figure 1. Characteristic features of CJD neuropathology consistent with the VV2 histotype A, B: Immunohistochemistry for PrP shows a deep laminar, perineuronal deposition pattern. C, D: in the basal ganglia, spongiform change is relatively prominent (C) and anti-PrP immunohistochemistry shows a strong synaptic deposition pattern (D). E-H: In the cerebellum, spongiform change is prominent in the molecular layer and there is a moderate reduction of granule cells (E). Single torpedoes are also observed (G). Immunohistochemistry for PrP reveals pathological synaptic deposits in the cerebellar cortex and coarse as well as plaque-like and periaxonal aggregates in the cerebellar white matter (H). Synaptic pathological PrP deposits were also detected in the brainstem including the locus coeruleus and solitary nucleus (not shown). In addition to the characteristic features of a spongiform encephalopathy, several fibrillar neuronal inclusions restricted to the locus coeruleus (LC) were identified in this patient. The locus coeruleus showed a moderate reduction of pigmented neurons and an accompanying gliosis. The inclusions were relatively large, partly eosinophilic and partly pale basophilic on H & E-stained sections and partly argyrophilic on Bielschowsky- and Gallyas silver impregnation methods (Figure 2). In contrast, by fluorescence microscopy, only a small fraction of the inclusions was weakly and irregularly stained by Thioflavin T, while the majority of inclusions was not stained by Thioflavin T.
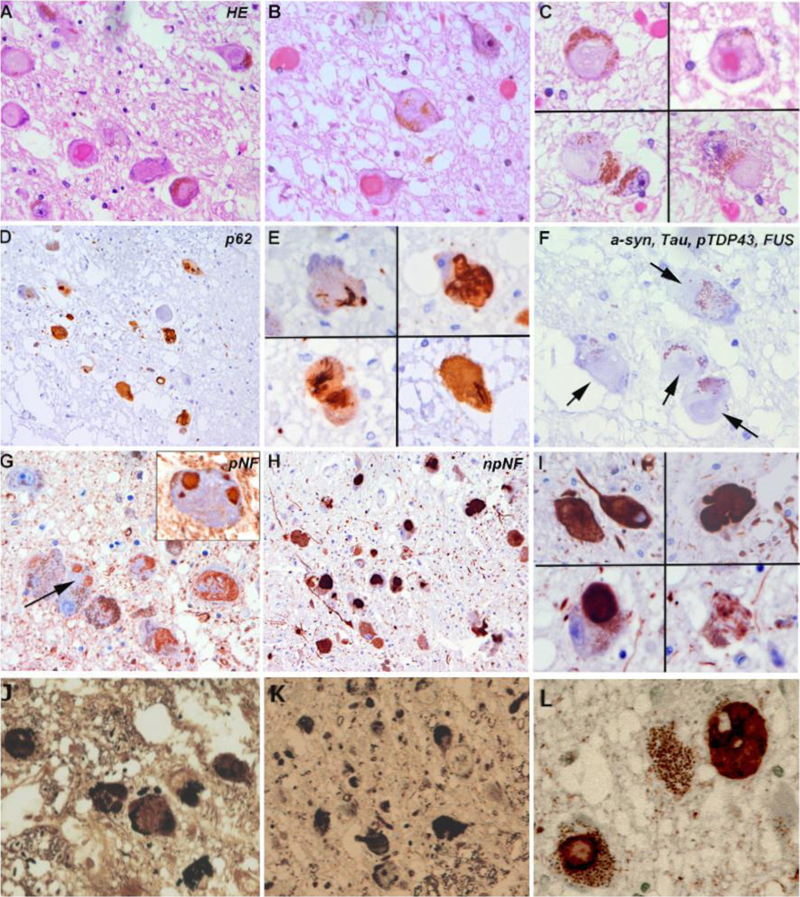
Figure 2. Microscopic features of intraneuronal cytoplasmic inclusions in the locus coeruleus A-C: Frequent filamentous intraneuronal cytoplasmic inclusions in the locus coeruleus after hematoxylin-eosin staining. Note the large size and the variable morphology of the inclusions that range from pale basophilic and fibrillar to dense hyaline and eosinophilic. D-E: p62 highlights the compact and filamentous nature of the large neuronal inclusions. F: Intraneuronal inclusions were negative for alpha-synuclein, tau, phosphorylated TDP-43 and FUS (arrows denote negative inclusion). G-I: In contrast, neuronal inclusions were positive for phosphorylated neurofilaments (SMI31, G, arrow points to a non-stained segment of a large fibrillar inclusion) and non-phosphorylated neurofilaments (SMI32, H, I). J-L: The inclusions were partly argyrophilic (J, K) and strongly positive for alpha-internexin (L). Immunohistochemically, the neuronal inclusions in the LC were immunoreactive for p62, an indicator of autophagic degradative activity and they were strongly immunoreactive for phosphorylated neurofilaments (BioLegend, clone SMI 31), non-phosphorylated neurofilaments (BioLegend, clone SMI 32) and alpha-internexin (Invitrogen, clone 2E3), a class IV neuronal intermediate filament. Although the histological and immunohistochemical profile of these neuronal inclusions strongly resembled that of neurofibrillary tangles, immunohistochemistry for phospho-tau (Thermo Scientific, AT8) remained negative. Immunostaining against alpha-synuclein (ajRoboscreen, clone 5G4), pTDP43 (Cosmobio, clone 11/9) and FUS (Sigma Life Science, clone G75241) were also negative. Interestingly, these fibrillar inclusions were not found in any other areas of the brain and were exclusively identified in the LC. Discussion We present a unique combination of sporadic CJD with striking neurofilamentous inclusions restricted to the locus coeruleus. This combination has not been described so far, and particularly, this type of neurofilament-bearing inclusions restricted to the locus coeruleus seems to be a singular finding. The significance of the inclusion bodies in this region is unclear and no obvious clinical correlate could be retrospectively extrapolated from the history of the patient, as symptoms were dominated by the rapidly progressive dementia as well as the brainstem and cerebellar symptoms of CJD. Of note, some important functions have been attributed to the locus coeruleus. The pigmented neurons in the pontine tegmentum have highly branched axon terminals that innervate many different areas of the CNS, including the neocortex, hippocampus, amygdala, thalamus, cerebellum and spinal cord and plays a decisive role in arousal, concentration and stress response as it is the most important source of norepinephrine in the CNS.12 The locus coeruleus seems to be prone to the development of certain types of intraneuronal inclusions, particularly neurofibrillary tangles and Lewy bodies, and its cells are affected in various neurodegenerative conditions.13 In Parkinson's disease or Lewy body dementia, alpha-synuclein aggregates in form of Lewy bodies and Lewy neurites occur in the locus coeruleus prior to the substantia nigra.14 The neuronal dysfunction of the locus coeruleus/subcoeruleus also plays a role, together with the gigantocellular nucleus of the formatio reticularis, in the development of rapid-eye movement behavior disorder (RBD), a disorder in which atonia lacks during REM sleep, leading to dream-enacting behavior with flapping, kicking and biting.15,16,17 RBD most often represents an early or even prodromal symptom of Parkinson's disease18 or Lewy body dementia and is less often encountered in multisystem atrophy. RBD is most frequently associated with alpha-synuclein pathology. Synuclein aggregates were however not observed in our patient. While we have no definite data on whether the patient had RBD or not, the large neurofilament inclusions in the locus coeruleus could theoretically be an additional non-LB related cause of RBD. Moreover, reduced cognitive flexibility in PD has been related to reduced neuromodulation of the prefrontal cortex from subcortical structures such as the locus coeruleus.19 These symptoms are however very difficult to assess retrospectively and separate from the development of CJD in our patient. Concerning prion diseases, experimental studies in mice have shown that the locus coeruleus, the solitary tract and the pre-Boetzinger complex are the first brainstem target areas of early prion deposition.20 Interestingly, the VV2 subtype of CJD also involves these brain regions, which all showed pathological PrP deposits in our case. Neurofilament inclusions are the hallmark of neuronal intermediate filament inclusion disease (NIFID), as described by Cairns et al.21 in patients with frontotemporal dementia. In these cases however, the morphology and particularly the extent of the inclusions are very different and affect also cortical areas, in particular fronto-temporal regions, basal ganglia, amygdala, hippocampus, brainstem including the substantia nigra, and cerebellum. The neurofilament inclusions were originally described as alpha-internexin positive and are currently considered as a subtype of FUS/FET proteinopathy.22 In our case, inclusions were positive for alpha-internexin, negative for FUS and in the majority negative for Thioflavin T. While Thioflavin T is a dye that binds to β-sheet rich structures, it may show variable affinity for different amyloid fibres. Whether the pale and irregular Thioflavin T staining of some intraneuronal neurofilament inclusions and the negative Thioflavin T staining in other inclusions reflect different fibrillation states and/or affinities of the aggregated protein to the dye is currently unclear. In summary, we report on a patient who, in addition to CJD, presented an additional and likely independent neuropathological alteration. The strict limitation of the large neurofilamentous inclusions to the locus coeruleus and their immunohistochemical profile do not match to any specific neurodegenerative condition but may be associated with subtle and slowly progressing neurological symptoms such as RBD or reduced cognitive flexibility. Declarations Conflicts of Interest Statement The authors declare that they have no conflicts of interest directly related to the topic of this article. Funding Statement The authors declare that they have not received any specific funding for this study. The Austrian Reference Center for Human Prion disease (ÖRPE) is supported by the Austrian Ministry of Health (Federal Ministry Republic of Austria for social affairs, health, care and consumer protection). Author Contributions OK and SW performed the autopsy; FL, OK, SW and EG conducted the neuropathological studies; SK and CS collected the clinical information, MRI and CSF data; FL and EG drafted the manuscript. All authors reviewed the manuscript. Acknowledgements We would like to thank the laboratory technicians at the division of neuropathology and neurochemistry, Medical University Vienna for their assistance. We deeply thank patients and their families. References 1. Hainfellner, J. A. et al. Coexistence of Alzheimer-type neuropathology in Creutzfeldt-Jakob disease. Acta Neuropathol 96, 116–122 (1998). DOI: https://doi.org/10.1007/s004010050870 2. Tousseyn, T. A. et al. Coexistence of Alzheimer-type neuropathology in Creutzfeldt-Jakob disease. Alzheimer’s & Dementia 4, T432–T433 (2008). DOI: https://doi.org/10.1016/j.jalz.2008.05.1285 3. Grau-Rivera, O. et al. Clinicopathological Correlations and Concomitant Pathologies in Rapidly Progressive Dementia: A Brain Bank Series. Neurodegenerative Diseases 15, 350–360 (2015). DOI: https://doi.org/10.1159/000439251 4. Iida, T. et al. An atypical case of sporadic Creutzfeldt-Jakob disease with Parkinson’s disease. Neuropathology 21, 294–297 (2001). DOI: https://doi.org/10.1046/j.1440-1789.2001.00407.x 5. Haraguchi, T. et al. Coexistence of Creutzfeldt-Jakob disease, Lewy body disease, and Alzheimer’s disease pathology: An autopsy case showing typical clinical features of Creutzfeldt-Jakob disease. Neuropathology 29, 454–459 (2009). DOI: https://doi.org/10.1111/j.1440-1789.2008.00964.x 6. Fernández-Vega, I. et al. Coexistence of mixed phenotype Creutzfeldt-Jakob disease, Lewy body disease and argyrophilic grain disease plus histological features of possible Alzheimer’s disease: A multi-protein disorder in an autopsy case. Neuropathology 35, 56–63 (2015). DOI: https://doi.org/10.1111/neup.12150 7. Rossi, M. et al. The characterization of AD/PART co-pathology in CJD suggests independent pathogenic mechanisms and no cross-seeding between misfolded Aβ and prion proteins. Acta Neuropathol Commun 7, 53 (2019). DOI: https://doi.org/10.1186/s40478-019-0706-6 8. Kovacs, G. G. et al. Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community-based autopsy series. Acta Neuropathol 126, 365–384 (2013). DOI: https://doi.org/10.1007/s00401-013-1157-y 9. Diagnostic Criteria | Creutzfeldt-Jakob Disease, Classic (CJD) | Prion Disease | CDC. (2021). https://www.cdc.gov/prions/cjd/diagnostic-criteria.html 10. Iwasaki, Y. The Braak hypothesis in prion disease with a focus on Creutzfeldt–Jakob disease. Neuropathology 40, 436–449 (2020). DOI: https://doi.org/10.1111/neup.12654 11. Parchi, P. et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Annals of Neurology 46, 224–233 (1999). DOI: https://doi.org/10.1002/1531-8249(199908)46:2<224::AID-ANA12>3.0.CO;2-W 12. Benarroch, E. E. Locus coeruleus. Cell Tissue Res 373, 221–232 (2018). DOI: https://doi.org/10.1007/s00441-017-2649-1 13. Zucca, F. A. et al. interactions of iron, dopamine and neuromelanin pathways in brain aging and parkinson’s disease. Prog Neurobiol 155, 96–119 (2017). DOI: https://doi.org/10.1016/j.pneurobio.2015.09.012 14. Braak, H. et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiology of Aging 24, 197–211 (2003). DOI: https://doi.org/10.1016/s0197-4580(02)00065-9 15. Dauvilliers, Y. et al. REM sleep behaviour disorder. Nat Rev Dis Primers 4, 19 (2018). DOI: https://doi.org/10.1038/s41572-018-0016-5 16. Boeve, B. F. et al. Insights into REM sleep behavior disorder pathophysiology in brainstem-predominant Lewy body disease. Sleep Medicine 8, 60–64 (2007). DOI: https://doi.org/10.1016/j.sleep.2006.08.017 17. Arnulf, I. REM sleep behavior disorder: Motor manifestations and pathophysiology. Movement Disorders 27, 677–689 (2012). DOI: https://doi.org/10.1002/mds.24957 18. Iranzo, A. et al. Neurodegenerative Disorder Risk in Idiopathic REM Sleep Behavior Disorder: Study in 174 Patients. PLoS ONE 9, e89741 (2014). DOI: https://doi.org/10.1371/journal.pone.0089741 19. Vazey, E. M. & Aston-Jones, G. The emerging role of norepinephrine in cognitive dysfunctions of Parkinson’s disease. Front Behav Neurosci 6, 48 (2012). DOI: https://doi.org/10.3389/fnbeh.2012.00048 20. Mirabile, I. et al. Identification of clinical target areas in the brainstem of prion-infected mice. Neuropathol. Appl. Neurobiol. 41, 613–630 (2015). DOI: https://doi.org/10.1111/nan.12189 21. Cairns, N. J. et al. Patients with a novel neurofilamentopathy: dementia with neurofilament inclusions. Neuroscience Letters 341, 177–180 (2003). DOI: https://doi.org/10.1016/s0304-3940(03)00100-9 22. Gelpi, E. et al. Phenotypic Variability Within the Inclusion Body Spectrum of Basophilic Inclusion Body Disease and Neuronal Intermediate Filament Inclusion Disease in Frontotemporal Lobar Degenerations With FUS-Positive Inclusions. J Neuropathol Exp Neurol 71, 795–805 (2012). DOI: https://doi.org/10.1097/nen.0b013e318266efb1
Copyright: © 2024 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |