|
|
||||||||||||||||||||||||||||||||||
|
Free Neuropathology 5:8 (2024) |
||||||||||||||||||||||||||||||||||
|
Review |
||||||||||||||||||||||||||||||||||
|
Neuropathology and epilepsy surgery – 2024 update |
||||||||||||||||||||||||||||||||||
|
Ingmar Blümcke |
||||||||||||||||||||||||||||||||||
|
Department of Neuropathology, University Hospital Erlangen, Germany |
||||||||||||||||||||||||||||||||||
|
Corresponding author: |
||||||||||||||||||||||||||||||||||
|
Submitted: 05 February 2024 |
||||||||||||||||||||||||||||||||||
|
Keywords: Brain, Histopathology, Seizure, Hippocampus, Neocortex, Dysplasia, Tumor |
||||||||||||||||||||||||||||||||||
|
Abstract Neuropathology-based studies in neurosurgically resected brain tissue obtained from carefully examined patients with focal epilepsies remain a treasure box for excellent insights into human neuroscience, including avenues to better understand the neurobiology of human brain organization and neuronal hyperexcitability at the cellular level including glio-neuronal interaction. It also allows to translate results from animal models in order to develop personalized treatment strategies in the near future. A nice example of this is the discovery of a new disease entity in 2017, termed mild malformation of cortical development with oligodendroglial hyperplasia in epilepsy or MOGHE, in the frontal lobe of young children with intractable seizures. In 2021, a brain somatic missense mutation of the galactose transporter SLC35A2 leading to altered glycosylation of lipoproteins in the Golgi apparatus was detected in 50 % of MOGHE samples. In 2023, the first clinical trial evaluated galactose supplementation in patients with histopathologically confirmed MOGHE carrying brain somatic SLC35A2 mutations that were not seizure free after surgery. The promising results of this pilot trial are an example of personalized medicine in the arena of epileptology. Besides this, neuropathological studies of epilepsy samples have revealed many other fascinating results for the main disease categories in focal epilepsies, such as the first deep-learning based classifier for Focal Cortical Dysplasia, or the genomic landscape of cortical malformations showing new candidate genes such as PTPN11, which is associated with ganglioglioma and adverse clinical outcome. This update will also ask why common pathogenic variants accumulate in certain brain regions, e.g., MTOR in the frontal lobe, and BRAF in the temporal lobe. Finally, I will highlight the ongoing discussion addressing commonalities between temporal lobe epilepsy and Alzheimer's disease, the impact of adult neurogenesis and gliogenesis for the initiation and progression of temporal lobe seizures in the human brain as well as the immunopathogenesis of glutamic acid decarboxylase antibody associated temporal lobe epilepsy as a meaningful disease entity. This review will update the reader on some of these fascinating publications from 2022 and 2023 which were selected carefully, yet subjectively, by the author. |
||||||||||||||||||||||||||||||||||
|
Introduction This review is a continuation of my 2022 update of interesting research papers and topics covering the search terms "neuropathology" and "epilepsy surgery" from 2021 [9], now discussing the 10 most interesting topics and/or papers published in 2022 and 2023. These search terms produced about 150 hits in PubMed in each year. The 'subjective' selection by the author is not an attempt at including all published papers or topics of the field and over this time period. Rather, I hope to highlight active areas of clinical and basic research with the potential to advance our concepts and understanding of disease pathomechanisms, e.g. how does a given 'molecular' pathomechanism translate into a lesional and epileptic phenotype. I will start with last year's "missing topic", i.e., the lack of published articles using artificial intelligence (AI) - based algorithms to support or supplement histopathology diagnosis of epilepsy-associated brain lesions. This chapter will also enlighten the discussion about the substitution of histopathologists by AI-driven technology as well as ongoing technical obstacles to successfully develop the field by histopathology experts rather than by industry-driven incentives. The first update to the internationally renowned FCD classification scheme, originally published in 2011 under the direction of the International League against Epilepsy (ILAE) and since then applied in epilepsy surgery units around the world as well as in research studies (cumulative citation index of > 1,199 in the Clarivate Web-of-science database as of January 2024), represents probably one of the most important papers published in the time period under review. The updated FCD classification has been already previewed in my 2022 update, however [9]. The 2022 update emphasized the impact of brain somatic mutations in lesional epilepsies such as Focal Cortical Dysplasia (FCD). Three international and collaborative research teams studied a series of 807 human brain tissues to unravel the genomic landscape of focal (lesional) epilepsies, which will be subsumed under item #2. These studies independently identified new epilepsy-related candidate genes, such as PTPN11, which belongs to the MAP-kinase signaling pathway and is well known from Noonan syndrome, a common RASopathy in children characterized by short height, congenital heart disease and often epilepsy, amongst many other clinical features. Interestingly, PTPN11 variants are most commonly detectable in temporal lobe epilepsy (item #3) and Ganglioglioma (GG) with adverse clinical outcome, respectively (item #6). The FCD update from 2022 also introduced a new disease entity, i.e. MOGHE, which was a topic of ongoing research in 2022 and 2023. Herein, I will review the clinical, genetic and histopathological assessments in a large, international cohort of 47 patients with MOGHE and SLC35A2 mutations (item #4). A first trial assessing D-Galactose supplementation in MOGHE patients with SLC35A2 mutations not being seizure free after surgery is the consequential next important step in the translation of our tissue research towards the implementation of personalized medicine and will be covered in the same chapter. The frequent localization of FCD ILAE Type 2 in the frontal lobe and of LEAT in the temporal lobe is another question not yet fully understood in the current literature. However, a recent Brain article tackled this question and delivered a fascinating hypothesis supported by the Allan Brain Atlas describing region-specific expression patterns of the mTOR protein to explain the impact of either loss-of-function or activating gene mutations in their associated signaling pathways (see also item #3). Temporal lobe epilepsy remains the most common form of focal epilepsy in adults and thus raises continuous interest. In the last four chapters, I will discuss a series of publications reporting expression of hyperphosphorylated tau in the epileptic condition as compared to neurodegenerative disorders (chapter #7). The impact of altered neurogenesis and gliogenesis in the dentate gyrus of the human hippocampus represents another hot topic (#8) as will the impact of hippocampal innate inflammatory gliosis (#9). These papers may redirect our research focus and interest on glial cells as important modulators if not initiators of epileptogenesis in the temporal lobe. My final topic addresses neuroinflammation and autoimmunity. The lymphocytic and microglial response to inflammatory pathomechanisms will always remain an important topic in the realm of focal epilepsy, and I do hope that my update will stimulate the readers' interest in this field.
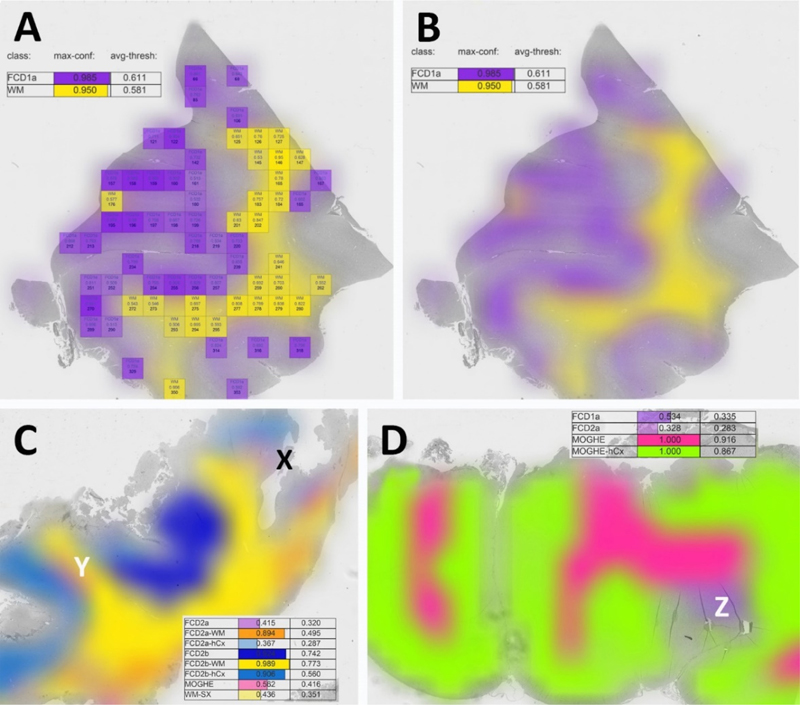
Topic 1: Which topic was missing in the 2022 update? AI-supplemented applications for routine histopathology review and diagnosis may be seen as the next era in Neuropathology. However, AI has not been used in the arena of epilepsy surgery yet and this was identified as a missing topic in my 2022 update review article [9]. Such a deep-learning based classifier for FCD was published in 2023 [43] and claimed to recognize most of the diagnostic entities defined in the 2022 FCD classification update [33]. The ResNet18 deep-learning algorithm was trained with extraordinarily large 1000 x 1000 µm digital tiles automatically extracted from whole slide images (WSI) of 414 manually annotated, H&E stained of FCD1a, FCD2a, FCD2b, mMCD and MOGHE samples (Figure 1). These were similarly processed to a series of 198 postmortem tissue blocks from 59 patients without neurological disorders including homotypic frontal, temporal and occipital areas and heterotypic Brodmann areas 4 and 17, entorhinal cortex and dentate gyrus. Overall, the algorithm's accuracy was specified as 98.8 % (F1 score = 0.82) in detecting 25 different anatomical regions and FCD categories. The classifier was also made temporarily available as an open access web application. Figure 1: Evaluation and visualization of a complete WSI slide obtained from epilepsy surgery
Figure 1. A: This is an example of the DL-based FCD classifier output as published by Vorndran et al. A digital slide (visible in grey in the background) was automatically segmented into a grid of tiles each measuring 1000 x 1000 µm. All tiles reaching the predefined confidence threshold (herein = > 0.9) can be visualized by a color-index map, e.g., purple or yellow. Summary values were also given on the upper left corner of the output screen. B: A blurring image filter is applied for the final output image hiding all information shown in A. Please note that the user can choose which of the available and trained FCD categories should be displayed. In this example, it is only FCD1a and white matter (WM). The sample was taken from a patient with histopathologically classified FCD1a [22]. C: A validation case example of a WSI obtained from a patient with histopathologically confirmed FCD2b and genetically confirmed pathogenic MTOR mosaicism. The lesion was detected and colored in dark blue for areas classified as FCD2b, light blue for neocortex associated with FCD2b, yellow for white matter associated with FCD2b. However, there were also areas recognized by the algorithm as FCD2a (X - light purple) and MOGHE (Y - pink). The algorithm's confidence score for these regions was low, however (< 0.415, 0.562, respectively). Such regions should then be reviewed at the light microscope by the responsible histopathologist. In this case, MOGHE (Y) was not confirmed histopathologically by the authors. D: A validation case example of a WSI obtained from a patient with histopathologically confirmed MOGHE with SLC35A2 mutation. Z points to a region with FCD1a as confirmed by the authors. Modified from [43] with permission from the author. Such digital slide suite projects will pave the way towards AI-supported histopathology diagnosis in years to come pending the clarification and resolution of ongoing obstacles in the field of whole slide imaging (WSI). Firstly, many parts of the world may not have access to a fast enough internet to upload such large digital WSI files, which can easily exceed multiple GByte (the range was described as 0.47-5.72 GByte per WSI in the current study). Secondly, a uniform laboratory standard of section thickness in the range of 4-5 µm and standardized H&E staining protocols must be applied to be compatible with the trained dataset of the AI algorithm. Thirdly and unfortunately most challenging, vendors of different WSI scanners implement their unique file formats [27]. There are open access WSI reading applications such as open.slide which can help to read and import different formats into an AI algorithm. However, any technical update of the file format or reading header by the vendor requires prompt adaptation by software stewards for any independent application. A solution to this problem would be the use of internationally defined DICOM standards, which already exists for WSI [27]. However, this has not been implemented by WSI vendors with the intention to control AI based histopathology slide suits as a walled garden laboratory solution wherein the WSI equipment and laboratory software must be purchased from the same company. In contrast, these applications should be openly available for research and managed by experts in the field to allow broad access to this technology irrespective of any country's or researcher's socio-economic background. Another open question remains, if AI-based histopathology classifier will replace the pathology expert in the near future. My personal belief is that AI will not replace the physician as the legal accountability of a signed pathology report for clinical patient management remains so high. Instead, such tools should help to prescreen routine cases. The application may then suggest a preselection of regions of interest, pending on an individually chosen level of the algorithm's detection sensitivity and which can be adapted by each reviewer (see Figure 2). Each reviewer must also determine if technical pitfalls have compromised the results, e.g., wrinkles, dust particles or other coloring errors, etc. These decisions will always remain with the responsible physician and cannot be replaced easily by any AI. However, this discussion will be ongoing in the community and my hope is that many world renowned experts will share their expertise in AI-based slide suites and classifier tools and which can be used open access around the world, in particular in those regions where training and experience in less broadly available. Figure 2: New genotype-phenotype associations in epileptogenic brain lesions
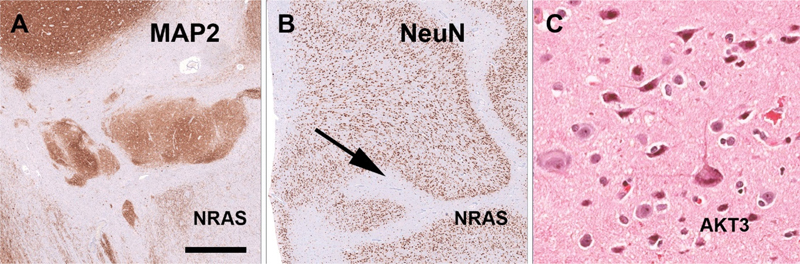
Figure 2. A-B: Two patients with a complex malformation of cortical development including heterotopias (A) and polymicrogyria (B) revealed brain somatic mutations in NRAS. C: A novel association was shown in another patient for a large chromosomal 1q duplication that included AKT3 and type FCD2a with hyaline astrocytic inclusions. Figure modified from open-access paper published under a Creative Commons license [30], with permission from the authors. Just to mention in this chapter that machine learning and AI tools were recognized and increasingly published already in the arena of epileptology [25], e.g., when virtual digital brain models can help to map a brain network in a given patient with epilepsy [24], when neuroimaging algorithms help to better identify structural brain lesions [44], or when predicting seizure outcome after epilepsy surgery in order to avoid unnecessary invasive diagnostic procedures for the patient [17]. Notwithstanding, the histopathology diagnosis often remains an important predictor in many of these AI-based applications. Topic 2: Deciphering the genomic landscape of epileptogenic brain lesions The search for genetic factors in lesional focal epilepsies is ongoing and pushed our knowledge of brain somatic mutations, chromosomal aberrations and epigenetic factors to a next level. I will review herein a series of three publications from 2022 and 2023 which comprehensively study brain tissues obtained from a total of 807 patients using different bioinformatic approaches. Chung et al. reported a multidisciplinary team effort including the FCD Neurogenetics Consortium and the Brain Somatic Mosaicism Network [13]. They retrieved 283 resected brain samples of patients with clinically and/or histopathologically validated malformations of cortical development (MCD) and identified a total of 69 mutated candidate genes. They had access to whole exome sequencing and targeted-amplicon sequencing platforms, and included in utero electroporation of the developing mouse brain to test the effect of their newly discovered genetic variants. Finally, single-nucleus RNA sequencing was employed to learn about the spatial gene expression patterns and enriched cell populations which may help to better understand the underlying pathomechanisms of MCD. The most commonly affected genes were, however, the already well-known candidates MTOR, PIK3CA, SLC35A2, TSC2, AKT3 and BRAF. Cheung et al. comprehensively studied their dataset from various angles. Firstly, their analysis of genes mutated in MCD highlighted four affected gene networks. The largest and most prominent cluster pointed to the well-known mTOR-MAP kinase pathway, in addition to a calcium dynamics cluster, a synapse cluster and a cluster aligned to gene expression. Then, the newly identified gene variants of PRAGA, GRIN2C and KLHL22 were electroporated into the developing mouse brain and revealed functional (migration) defects resembling that of a cortical malformation. Thirdly, their genotype-phenotype correlation highlighted an association of FCD ILAE Type 2b and 2a with mTOR rather than MAP-Kinase pathways. This association has been now consistently reported [23] and will be further discussed in the chapter below. Finally, they studied single-nucleus spatial transcriptomics of mutated genes obtained from patient samples versus controls, which indicated a critical role in excitatory neurogenic cell pools during brain development, and further supported the neurobiological basis for enhanced neuronal hyperexcitability in the epileptogenic brain. Of note, all SLC35A2 mutated cases were histopathologically classified as FCD 1 and the BRAF cases as FCD 3. This is likely a histopathology-disagreement error frequently encountered in the histopathology work-up of epilepsy surgery tissue samples and this discussion will be continued below. In fact, re-review of published cases carrying a SLC35A2 gene mutation were frequently re-classified as MOGHE [6, 10, 12]. The BRAF mutations described by Cheung and colleagues most likely represents ganglioglioma tumor cell infiltration into surrounding brain tissue, which is often regarded as an associated FCD ILAE Type 3b. As another example for disagreement in histopathology diagnosis, however, a recently published paper claimed a new diagnostic entity with BRAF mutated cells invading/infiltrating the hippocampus [29]. Such cases have been seen also in our routine practice but regarded rather as remnants of a ganglioglioma infiltrating the hippocampus. Cheung and coworkers also briefly mention PTPN11 as new lesional epilepsy gene. Two other studies have made the same observation but at a much larger frequency of affected samples and diagnostic emphasis. In the second paper, López-Rivera and coworkers examined a total of 474 new samples not previously reported in other studies [30]. The surgical tissues were selected from four different centers in Germany and the US. Histopathology diagnosis covering the spectrum of MCD (n = 223), LEAT (n = 154) and Hippocampal Sclerosis (n = 97) were included. They reported nineteen genes of interest and reported 153 samples carrying at least one single nucleotide variant, copy number variant or loss of heterozygosity affecting 31.4 % of the entire cohort, 7.2 % of HS, 31.8 % of MCD and 51.9 % of LEAT. These numbers remain low despite their effort to use deep sequencing rates at a mean depth of > 350 x. A novel association was histopathologically established for PTPN11 with low-grade epilepsy-associated brain tumors (Ganglioglioma), NRAS with a complex MCD including polymicrogyria, and AKT3 with FCD 2a and hyaline astrocytic inclusions (Figure 2). In addition, chromosomal losses for germline variants recognized for TSC2, DEPDC5 and PTEN were most compatible with the previously proposed second hit hypothesis to also play a role in the etiopathology of MCD [36]. The authors then concluded to design MCD-specific panels for clinical routine testing of surgical samples to support clinical diagnosis but also enable targeted treatment in those patients that failed successful postsurgical seizure control. A third, smaller study from the US enrolled surgical brain tissue samples obtained from 50 children and used whole exome and RNA sequencing [7]. Overall, 56 % of their patients could be explained by genetic findings, including the SLC35A2 gene and mTOR pathway genes in MCD, and MAP-Kinase pathway genes in tumors. Amongst the MAP-Kinase pathway genes, the authors were the first to report PTPN11, which was detected in two samples including FCD IIIa and IIId. They also reported SLC35A2 genotypes in two patients with the histopathology diagnosis of FCD Ic, but these histopathology phenotype-genotype correlations may not stand an independent second review. As mentioned above, an independent re-review of previously published SLC35A2 altered samples identified the 'new disease entity' of MOGHE to be the underlying pathology rather than any FCD or mMCD subtypes [6, 12]. The same conclusion is drawn from the paper of Carmen Barba reviewing 47 patients with MOGHE as discussed below (#4). Topic 3: A new hypothesis about regional mTOR gene variant accumulation in the frontal lobe and MAP-Kinase gene variant accumulation in the temporal lobe The genomic landscape of focal lesional epilepsy gets better recognized as large enough patient series are examined. However, it remained a hitherto open question why some lesions and their associated genetic alterations occur overwhelmingly frequent in certain brain localizations. As a prominent example, FCD ILAE Type 2 lesions carry constitutively activating mTOR pathway gene variants and occur most frequently in the frontal lobe, same as do SLC35A2 altered MOGHE. In contrast, GG and other low-grade epilepsy associated brain tumors (LEAT) overwhelmingly often carry activating MAP-RAS-RAF Kinase signaling pathway gene variants (e.g., BRAF or PTPN11) and occur predominantly in the temporal lobe. A recent Brain publication from Macdonald-Laurs and coworkers offered an intriguing answer to that question [31]. By studying the clinical, pathologic and genetic landscape of their cohort of 85 patients with FCD at the bottom-of-sulcus (BOSD), which is a peculiar FCD Type 2 variant specifically highlighted also in the international FCD classification update of 2022, they reported that 62 % of their cohort presented in the frontal lobe, 18 % in the parietal lobe, and 14 % in the insula, whereas the temporal and occipital lobes were rarely affected (5 % and 1 %, respectively). Only pathogenic mTOR pathway gene variants were detected and occurred in 60 % of their patient cohort. Then, they compared the regional frequency of the BOSD with the mTor protein expression pattern published in the open access online Allen Human Brain Atlas (Figure 3). Intriguingly, mTOR is less dominantly expressed in the frontal lobe compared to temporal or occipital lobes suggesting that a gain-of-function alteration of mTOR signaling, e.g., MTOR activating mutations or inactivating DEPDC5 mutations/losses, would have had a much higher impact in the frontal lobe where these genes are not naturally highly expressed. Notwithstanding, this hypothesis needs to be studied further. An intriguing study would be to search for pathogenic mutations in mTOR or MAP-Kinase pathway genes in other brain regions not affected by a pathology lesion of the same patients or in the general population. If one can prove their presence it could well support the functional impact in generating a structural lesion only when their signal gets abnormally high during the neurodevelopmental process. Figure 3: Brain expression of mTOR suggests regional vulnerability for FCD 2
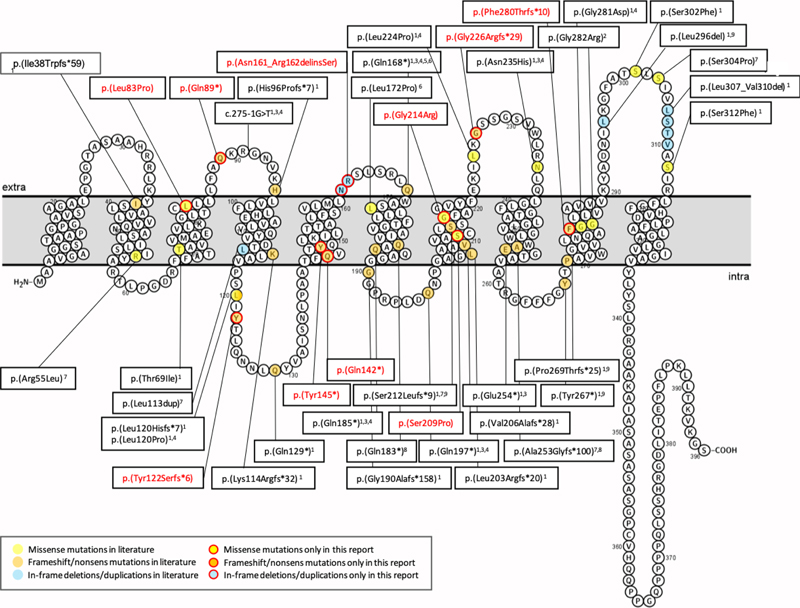
Figure 3. Schaefer parcellation of the lateral and medial surface of the left hemisphere showing (A) the number of bottom-of-sulcus-dysplasia (BOSD) with at least 10 % overlap with each parcel, and (B) relative mTOR expression from the Allen Human Brain Atlas. Note that privileged regions of BOLD associated with low expression of mTOR. Figure modified from open-access paper published under a Creative Commons license [31], with permission from the authors. A same regional accumulation of activating mutations of the MAP-Kinase pathway, such BRAF or PTPN11 have been identified in the temporal lobe, as recently published by Khoshkhoo and coworkers [26]. They studied 105 individuals with mesial temporal lobe epilepsy (MTLE) compared to 30 neurotypical controls. They detected 11 pathogenic somatic variants in hippocampal tissue neurosurgically resected in their patient cohort but none in their controls. These variants included PTPN11, SOS1, KRAS, BRAF and NF1, all of which are predicted to constitutively activate the MAP-Kinase signaling pathway and which might contribute to the pathogenesis of sporadically occurring, drug-resistant MTLE. In other words, not only MAP-Kinase associated low-grade brain tumors but also hippocampal sclerosis accumulate MAP-Kinase activating pathogens and may contribute to its high epileptogenic susceptibility. Topic 4: New diagnostic disease entities: I. MOGHE The clinico-pathologic diagnosis of MOGHE as a new disease entity in patients with focal, drug-resistant early onset (frontal lobe) epilepsy was first described in 2017 [37]. Since then, scientific and clinical interest has continuously grown towards a better understanding of MOGHE. In 2018, a novel brain somatic loss-of-function mutation of the galactose-transporter SLC35A2 gene was identified in epilepsy surgery brain tissues [38, 45]. This was confirmed in 2019 [5] and all three publications assigned the SLC35A2 mutation to variable histopathology phenotypes, such as mMCD, FCD Type 1 or non-lesional samples. Two papers concluded in 2021, however, that the SLC35A2 mutation is specifically associated with MOGHE and found in about 50 % of reported patients [10, 12]. This enduring uncertainty stimulated an international consortium of 13 epilepsy centers around the world led by Dr. Barba to review the neuropathology, clinical features, and postsurgical outcome in a total of 47 patients previously described to carry brain somatic SLC35A2 mutations [6]. The microscopic re-review of all available surgical specimens identified MOGHE in 94 % of their patient series, leaving very limited room to assign the SLC35A2 mutation to any other disease or FCD entity. As a matter of fact, the diagnosis was inconclusive in 3 patients because of insufficient surgical sampling (6 %). Twenty-nine cases were previously published with a diagnosis of FCD Type 1, mMCD or non-lesional, which needs reconsideration and which also highlights the difficulty to achieve agreement in the histopathological diagnosis of epilepsy surgery brain samples as was already discussed above. Interestingly, they observed two clinical phenotypes of MOGHE. The majority of their patients (n = 39; 83 %) had early epileptic encephalopathy with a seizure onset range from 3 month to 3.5 years and severe to moderate intellectual disability. The second and smaller cohort of eight patients (17 %) could be characterized as drug-resistant focal epilepsy of young to adolescent (later) onset associated with almost normal cognitive function. Another interesting observation was the lack of any hot spot mutation across the SLC35A2 gene (Figure 4). All mutations were predominantly distributed across the nine transmembrane domains. Finally, there was no association between the genetic variant or variant allelic frequency (VAF), which ranged from 1.4 % - 52.6 % in all samples (mean VAF = 17 %), with the clinical phenotype or the postsurgical outcome. Indeed, seizure freedom was achieved in 30 patients (63.8 %, Engel Class I), and favorable outcome was significantly associated with shorter disease duration and complete resection. These numbers dramatically changed from the first published results in 2017, where only 33 % of patients were reported to achieve postsurgical seizure control. Partial resection due to unknown lesion boundaries were the most likely explanation in this early study. The challenge to anatomically define MOGHE's lesion borders remains actually, which may open possible avenues for pre- or perioperative online sequencing methodologies using the real-time, direct sequencing technology, e.g. see [42], from intraoperative specimens or depth electrodes obtained from presumptive lesion borders. It is yet unknown, however, if the low variant frequency in these lesions and lack of hot spot mutations can be resolved by the technology. Figure 4: Membrane topology modeling of currently known SLC35A2 mutations in MOGHE
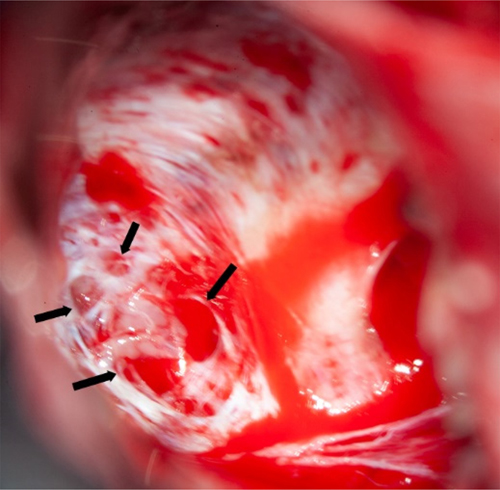
Figure 4. Schematic representation of variants reported by [6]. Novel variants are labeled in red, variants already described are labeled in black. Membrane topology was predicted using the Protter online tool41 (P78381-SLC35A2_HUMAN). Figure collected from open-access paper published under a Creative Commons license, with permission from the authors. In 2023, another small study described five new patients with MOGHE confirming the predominance of frontal lobe epilepsy and epileptic encephalopathy phenotypes in patients histopathologically diagnosed as MOGHE [18]. However, this study has not applied genetic testing for brain somatic mutations. As reported above, all patients responded favorably to extensive frontal lobe resections, despite widespread epileptic activity recorded by surface and intracranial EEG pre- and postoperatively. The authors concluded, therefore, that an epileptic encephalopathy phenotype in the first years of life should not discourage a surgical treatment option. An additional recently published paper from a European consortium of epilepsy surgery centers led by Dr. Angel Aledo-Serano from Madrid, Spain sheds further light on MOGHE. Twelve patients with histopathologically confirmed MOGHE were included in their postsurgical clinical trial attempting to test the benefit of D-galactose supplementation when patients were not seizure free after surgery. So far, research evidence reached the conclusion that at least 50 % of MOGHE patients suffer from a brain somatic loss-of-function mutation in the galactose transporter gene SLC35A2, which delivers galactose into the endoplasmic reticulum and Golgi apparatus, thereby glycosylating sphingolipids into functionally active protein epitopes. One therapeutic option to circumvent the lack of slc35a2 protein would be to push alternative transporter pathways by offering vast amounts of galactose substrate to the patient. This treatment strategy was tested in 2020 in young children with the germline variant of SLC35A2 deficiency, i.e. SLC35A2-congenital glycosylation disorder characterized by epileptic encephalopathy, developmental disability, growth deficiency, dysmorphism as well as high mortality rates within the first years of life [47]. Their hypothesis was that galactose supplementation partially overcomes the Golgi UDP-galactose deficiency and improves galactosylation [47]. In addition, oral galactose supplementation was well tolerated showing promise as dietary therapy. Aledo-Serano aimed, therefore, to evaluate the effects of D-galactose supplementation also in patients with histopathologically confirmed MOGHE, with uncontrolled seizures or cognitive impairment and epileptiform activity at the EEG after epilepsy surgery as inclusion criteria to their clinical trial (NCT04833322). Patients were orally supplemented with D-galactose for 6 months in doses up to 1.5 g/kg/day and monitored for seizure frequency including 24-h video-EEG recording, cognition and behavioral scores, before and six months after treatment. Positive response was defined by more than 50 % improvement of seizure frequency and/or cognition and behavior. Twelve patients aged 5-28 years were included from three different centers. Neurosurgical tissue samples were available in all patients and revealed a brain somatic variant in SLC35A2 in six patients (not-present in the blood). After 6 months of supplementation, D-galactose was well tolerated with just two patients presenting abdominal discomfort, solved after dose spacing or reduction. There was a 50 % reduction or higher of seizure frequency in 3/6 patients, with an improvement at EEG in 2/5 patients and a global responder rate of 6/6 in SLC35A2-positive patients. One patient became seizure-free. An improvement of cognitive/behavioral features was also observed. In contrast, only three out of nine responders carried the SLC35A2 wildtype gene and all non-responders were SLC35A2 wildtype. Their results suggested that supplementation with D-galactose in patients with MOGHE is safe and well tolerated and, although the efficacy data warranted larger studies, it might build a rationale for precision medicine after epilepsy surgery. Topic 5: New diagnostic disease entities: II. Encephaloceles Encephaloceles (ENC) are protrusions of the neocortex and its meninges expanding into the diploic space of the skull bone (Figure 5), i.e. the medullary cavity, leaving the outer skull table intact [4]. ENC can occur in any location, most commonly in the frontal or parietal region [4]. They can develop congenitally, as will be discussed herein, which needs to be separated from a traumatic, neoplastic, inflammatory or iatrogenic origin. Increased intracranial hypertension, e.g., empty sella or optic nerve sheath dilatation, female gender and obesity have been repetitively reported as risk factor for ENC when occurring without a clear etiology. In patients with drug-resistant temporal lobe epilepsy (TLE), ENC are increasingly recognized in the temporo-polar and/or basal region of the brain. These are likely congenital ENC and can vary in size from mm to cm, vary in number up to more than 10 in a focal region and affect either one or both hemispheres. Very small ENC are often seen only during surgery as they escape the current resolution scale of clinical routine MRI. A recent paper from Di Giacomo and coworkers looked at electro-clinical, neuroimaging and histopathology features of their series of 12 patients with ENC [16]. All patients were retrospectively identified by reviewing 3T MRI scans. There were no exact numbers of ENC given per patient and data from their patient table suggested one MRI-visible ENC was present in either one (n = 6 patients) or in both hemispheres (n = 6 patients). However, eight patients underwent neurosurgical resection of only one hemisphere following a careful neurophysiological video-EEG assessment of the seizure origin. All patients were postoperatively seizure free (Engel class I). The surgical tissue specimens allowed for a systematic histopathological analysis, which revealed focal distortion of Layer I, white matter extending into the neocortex and/or an altered gyral profile. Considering the difficulty to anatomically reconstruct such small protrusions in a surgical sample, their analysis comprehensively describe ENC as a common histopathology entity to be recognized and diagnosed within the lesional spectrum of epilepsy surgery. I found their description of a focal distortion of neocortical Layer I quite interesting, as it reliably describes for the first time a disease condition with horizontally oriented cortical layer abnormalities compatible with FCD ILAE Type 1b. The FCD 1b subtype is neither well studied nor characterized yet and some people even question their existence due to the lack of a characteristic and consistent clinico-pathological phenotype. Nevertheless, the authors did not claim such a FCD classification and we need to await further studies to better understand its likely neurodevelopmental origin and pathogenesis. A genetic or epigenetic characterization would also be helpful to assess their origin and specificity as new disease entity. Notwithstanding, the clinical history available in their presented patient cohort may add to the disease description as a majority revealed MRI positive intracranial hypertension (9 out of 12 patients), later onset of seizures compared to a control group of 26 patients with non-ENC related TLE, and more frequent psychiatric comorbidity. Figure 5: Intraoperative snapshot demonstrating multiple encephaloceles in the human temporal lobe
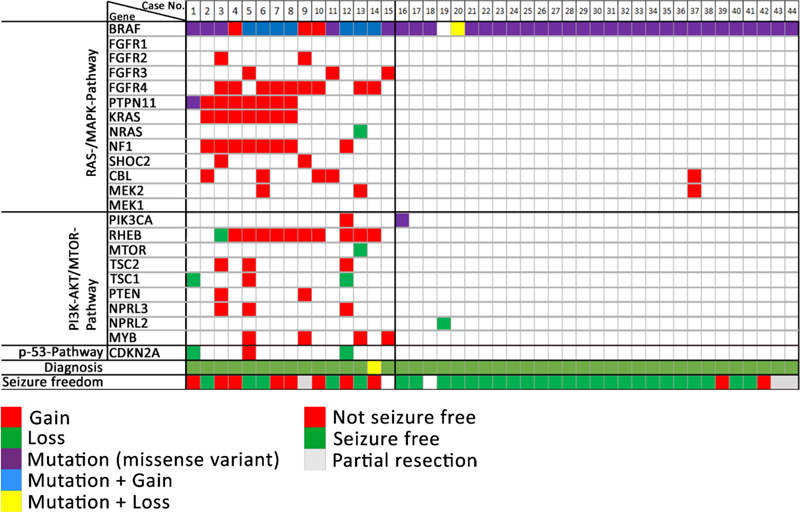
Figure 5. The intraoperative vision angle is directed towards the basis of the temporal pole in a 23-year old patient with left TLE. There were several larger and smaller holes in the skull bone as indicated by the arrows, some of which were too small to be preoperatively detected by MRI. All clefts were occupied by cortical brain tissue protrusions. The exact histopathology and/or genetic nature of these encephaloceles need further clarification. Image not previously published and kindly provided Prof. S. Brander, Neurosurgery Dept. at Klinikum Fürth, Germany. Topic 6: What did we learn in 2022 and 2023 about LEAT Patients suffering from low-grade epilepsy-associated brain tumors (LEAT) have a high propensity to become seizure and drug free after epilepsy surgery [28]. This is a promising observation factoring into every day's patient management and counseling. The question remains, however, why up to 30 % of patients with LEAT do not achieve sufficient seizure control postsurgically. Unfortunately, this measure is not recognized in the WHO brain tumor classification scheme and does, therefore, not play a role in our current neurooncology-focused literature of low-grade brain tumors. This contrasts a recently published work from Hoffmann and coworker presenting their series of 72 LEAT samples submitted to whole exome sequencing [21]. The same patients were presented also in the work of López-Rivera described above (see topic #2). There were eight patient samples, in which the aforementioned PTPN11 gene was altered, mostly by a gain of the long arm of chromosome 12. These eight patients also showed chromosomal gains affecting other genes of the MAP-Kinase pathway, e.g., SHOC2, BRAF, KRAS, NF1 or FGFR4, representing a much more complex genotype than usually seen in the group of GG. Another seven patients showed a similarly complex genotype including several genes of the MAP-Kinase and mTOR signaling pathway (Figure 6). Compared to their group of 29 patients with a GG and a BRAF V600E mutation, the group of GG with a complex genotype had a much lower seizure control two years after surgery (38 %). This contrasted the 85 % of successful postsurgical seizure control in the BRAF altered group of GG. Histopathology analysis of this cohort revealed additional features of what the authors describe as atypical features in GG (analogue WHO CNS grade 2), e.g., subarachnoidal tumor spread of predominantly astroglial and dysplastic neuronal phenotypes, as well as a consistent combination of CD34 and p16 immunoreactivity. Finally, a new unsupervised DNA methylation cluster analysis was presented including more tumor samples from previously published cohorts readily separating between GG, dysembryoplastic neuroepithelial tumors (DNT), low-grade glioma MYB altered, and pleomorphic xanthoastrocytoma (PXA). There was a fifth cluster, however, including all of the PTPN11 altered GG and even more tumor samples, some of which also had an adverse postsurgical outcome (one patient died from SUDEP as seizures could not be controlled). Figure 6: Oncoplot of WES and CNV detection in ganglioglioma with clinically adverse outcome
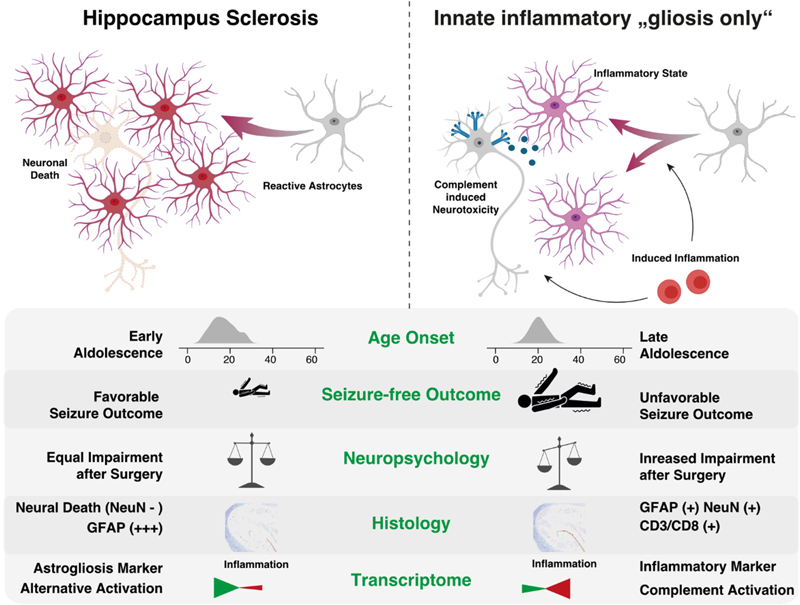
Figure 6. Columns 1-44 represent patient samples submitted to WES. These data were intentionally classified into three groups of patients: GG with complex genetic variants (cases 1-15), GG with non-complex genetic variants (cases 16-44), and GG without any detected genetic variants (cases 45-72, not shown). Diagnosis: Histopathology review confirmed the diagnosis of ganglioglioma (green tiles) in 43 cases. Lack of seizure freedom (red tiles) was explicable by incomplete resection only in cases #9, #42, and #43 (greyish tiles). Outcome data was missing in cases #15 and #18 (white tiles). Figure collected from open-access paper published under a Creative Commons license, with permission from the authors. These results prompted the authors to suggest that the WHO panel may use a different strategy when classifying LEAT, at least reconsidering a grading scale which re-introduces WHO CNS grade 2. As a reminder, previous editions of the WHO brain tumor classification included even anaplastic variants of GG. Along these lines, Reinhard and coworkers recently published their study addressing an internationally collected cohort of 54 anaplastic ganglioglioma, often including epilepsy, tumor relapse and second surgeries in the patient's histories, by using the existing Heidelberg DNA methylation classifier [35]. The majority of tumors designated as anaplastic gangliogliomas in the original histopathology report resolved into other WHO diagnoses, most commonly PXA (30 %) and glioblastoma, IDH wildtype (20 %). Only ten of their tumor samples were not assignable to a CNS WHO diagnosis. It remains to be shown, however, if atypic or anaplastic ganglioglioma do not exist at all as a category defined also by DNA methylation. The work of Hoffmann et al. approved such an atypic category, but using a different bioinformatic programming pipeline [21]. It is the availability of enough and clinically well annotated tumor samples covering the entire landscape of LEAT that will make the difference. The issue of a lower tumor cell content in LEAT represents an additional challenge to be also addressed in any DNA methylation brain tumor classifier. These conflicting data may also pave the way towards new approaches to classify brain tumors, e.g., using the real-time, direct sequencing technology [42], and that a single histopathology consultant may need to apply different systems to obtain a sound picture of the underlying tumor diagnosis. That clinical considerations play a major role in LEAT and that individual approaches in developing DNA methylation classifier can also be concluded from a paper of Stone and coworkers [40]. They asked the question if DNA methylation is helpful to synergize the classification of LEAT into histopathology and neuroimaging similar subgroups and constructed a methylation-based support vector machine model for their prediction. They extended a previously published series now including 83 LEAT samples and comprising glio-neuronal tumors of two major phenotypes, i.e., GG and DNT [39]. These tumor entities indeed segregated into two groups. At the histopathological level, prominent astrocytic or oligodendrocyte-like components, dysplastic neurons or a specific glioneuronal element were important discriminators between both groups. At the neuroradiological level, the location, margin definition, enhancement and T2 FLAIR-rim sign were successful discriminators. In a validation round their AI-based algorithm classified 22/23 samples correctly. The authors concluded the importance and superiority of an integrated diagnostic approach using histopathology, molecular data but also clinical information as our current diagnostic criteria inadequately reflect glioneuronal tumor biology and leave a proportion unresolvable. These recent papers continue to pose questions how to reliably classify and grade LEAT, and agree on the terminology use. Continued disagreement will affect any interpretation of published research and also obstructs a better prediction of their clinical behavior beyond being regarded as benign WHO CNS grade 1. Topic 7: Tauopathies in mesial temporal lobe epilepsy There is increasing research interest in tauopathies related to temporal lobe epilepsy. Based on the original observations of abnormal tau phosphorylation in drug-resistant epilepsy [48] that aggregates into neuropil threads (NT) or neurofibrillary tangles (NFT) in brain biopsies surgically resected from patients with drug-resistant epilepsies, there is an ongoing discussion about precocious aging in TLE patients as a result from similar pathomechanisms observed in Alzheimer's disease. This claim was not only spectacular at that time but may bridge the open question of ongoing and aggravating memory decline in drug-resistant TLE patients. However, available research data does not fully support the hypothesis anymore as will be shown in a small compilation of three recently published papers. Aroor and coworkers addressed the possibility that cognitive decline in epilepsy may be associated with mechanisms typical of Alzheimer's disease (AD) [3]. They determined the abundance of phospho-tau and Aβ proteins in association with cognitive function in 12 cases of drug-resistant epilepsy. Indeed, they could observe a robust presence of tauphospho-related NT (Ser202/Thr205) and NFT pathology, as well as Aβ deposits, and phosphoS6 (phosphorylation of S6 at Ser240/244 or Ser235/236 indicating an activation of the mTOR complex) in the epileptic samples. They found no significant correlation, however, between tauphospho (Thr205; Thr181), Aβ, or mTOR markers with cognitive scores. These findings still support the existence of hyperphosphorylated tau protein and Aβ deposits in patients with drug-refractory TLE, but their relation to cognitive decline remains uncertain and requires further investigation. Concepcion and coworkers studied the progressive dysregulation of tau phosphorylation in a rat pilocarpine status epilepticus (SE) model of TLE [14]. They measured tau expression at two months and four months after SE. Total tau levels were modestly reduced compared to naïve controls at two months post-SE, but there was no significant reduction in S202/T205 phosphorylation. In the whole hippocampal formation from four month post-SE rats, total tau expression had reverted to normal, but there was a significant reduction in CA1 and CA3 for S202/T205 tau phosphorylation. No change in phosphorylation was seen at the T181 and T231 tau loci. In somatosensory cortex, outside of the seizure onset zone, no changes in tau expression or phosphorylation were seen at the later time point. The authors concluded that total tau expression and phosphorylation did not show hyperphosphorylation at the three AD canonical tau loci tested in their animal model of TLE. Instead, the S202/T205 locus showed progressive dephosphorylation. This suggested that changes in tau expression may play a different role in TLE than in AD. Further studies are needed, therefore, to understand how such tau changes may impact neuronal excitability in chronic epilepsy. Witt and coworkers approached this interesting topic from a neuropsychology standpoint claiming that the etiology of cognitive deficits in epilepsy is really multifactorial, the neuropathology of MTLE rather multifaceted and beyond that of justifying the study of hyperphosphorylated tau [46]. Indeed, the bidirectionality between epilepsy and dementia is of high interest, also from the epileptological perspective. However, there are many etiologies in TLE that were associated with cognitive decline in epilepsy, as histopathology findings in MTLE samples reported hippocampal sclerosis, dysplastic lesions, and neurodevelopmental neoplasm as major disease categories. Last but not least, anti-seizure medication can also have adverse effects on cognition. These authors concluded that the neuropsychology and neuropathology of MTLE is actually more complex than recently postulated [49]. The suggested model that AD-TLE represent a specific disease subtype needs further validation to confirm the role of hyperphosphorylated tau in epilepsy patients with and without AD considering age and age at epilepsy onset as potential moderator variables. Topic 8: 'Hippocampal innate inflammatory gliosis only' in pharmacoresistant TLE Hippocampal Sclerosis (HS) remains the most common structural brain lesion of all focal epilepsies and is the hallmark of Temporal Lobe Epilepsy (TLE). Histopathology patterns vary, however, in this cohort of affected individuals and the ILAE has developed a 3-tiered classification scheme based on the pattern of segmental neuronal cell loss in the CA regions [11]. There has always been a fourth category of 'no-HS, gliosis only', however. This category represented almost 20 % of the entire cohort of TLE patients with surgical resection of the hippocampus and clinical histories were described as slightly different from the HS cohort, i.e. later seizure onset and later age at surgery, respectively. Grote and coworkers have addressed this challenging category of 'no-HS, gliosis only' with a comprehensive and innovative clinico-pathological and molecular study design [19]. They reviewed a cohort of 627 patients with drug-resistant TLE submitted to hippocampal resection. Seventy samples showed 'no-HS, gliosis only' at the histopathology examination (11 %). Indeed, a careful review of the clinical charts confirmed a significantly later onset of seizures when compared to HS (16.3 vs. 12.2 years, p < 0.005), and a significantly worse rate of seizure freedom after surgery (43 % vs. 68 %, p = 0.0001). More interestingly, however, was their RNA sequencing analysis comparing no-HS vs. HS samples, which revealed a distinct transcriptional program visible only in the no-HS group and which suggested an innate inflammatory response of reactive astrocytes. Notwithstanding, reactive astrocytes are the cellular correlate of 'gliosis only', histopathologically characterized by a diffuse astrogliosis pattern lacking restricted segmental focality and which is, therefore, poorly controllable by surgery. Poorer postsurgical outcome in this cohort should alert any physician, therefore, when red flags occur during the presurgical evaluation, e.g., clinical and demographic features such as later disease onset, negative MRI findings or bilateral epileptogenicity patterns (Figure 7). A more adequate treatment algorithm should rather address the innate inflammatory tissue response, but these may not be readily available yet. However, the same group has published their results of fingolimod treatment in an experimental TLE model targeting the innate inflammatory gliosis in 2019 [34]. Fingolimod targets sphingosine-phosphate receptors (S1PRs) and revealed robust anti-convulsive activity in kainate-induced SE mice. In addition, it had neuroprotective and anti-gliotic effects and reduced cytotoxic T cell infiltrates suggesting repurposing of fingolimod (previously known as suitable drug in Multiple Sclerosis treatment) as novel therapeutic option in focal epilepsies. It is hoped, therefore, that patients with no-HS can be identified early on during the presurgical evaluation phase to carefully consider all therapeutic options. Figure 7: Graphical summary of the differences between hippocampal sclerosis and Innate inflammatory 'gliosis only' (I²GO)
Figure 7. The authors claim that I²GO constitutes a distinct MTLE syndrome with characteristic clinical and pathological features. I²GO is less amenable by surgery and bears a greater hazard for postoperative neuropsychological deterioration. 'Gliosis only' is the neuropathological hallmark of I²GO and shows a unique transcriptional signature marked by an astrocyte-mediated chronic inflammation pattern. Figure collected from open-access paper published under a Creative Commons license, with permission from the authors. Topic 9: Altered adult neurogenesis and gliogenesis in patients with mesial TLE The impact of gliogenesis and reactive gliosis in epileptogenic human tissue has also been addressed in a seminal paper published by Ammothumkandy and coworkers [2]. They used immunofluorescence microscopy in human surgical hippocampus obtained from 19 MTLE patients, performed neuronal stem-cell cultures and multi-electrode array recordings of ex vivo hippocampal slices to address the hitherto open question if and how aberrant neurogenesis and gliogenesis play a role in seizure initiation and progression. This has been repetitively reported in rodent animal models but needs further confirmation in the human disease condition. As reported in previous studies (see below), the authors confirmed a decline of neurogenesis in TLE patients when their disease had a longer duration. These data were obtained from fixed tissue specimens as well as in vitro from neural stem cell cultures. Colocalization of Dcx+ and Prox1+ immunoreactivity was used to recognize immature dentate granule cells and by comparing biopsy tissue with age-matched postmortem controls. In contrast, gliogenesis remained persistently high even in the chronic epileptic condition. Immature astroglia were characterized by Dcx+/Prox1- and GFAP+ immunoreactivity compared to their mature counterpart being only GFAP+ immunoreactive. 400 µm thick slices of the human hippocampus were then used for multi-electrode array recordings followed by immunofluorescence stainings of the aforementioned cell marker sets. Based on these findings they classified tissue regions as active or non-active. Neurogenesis was convincingly proven only in neurophysiologically non-active tissues but not in tissue with epileptiform activity. They concluded from this set of experiments that newborn neurons do not contribute to epileptiform activity at chronic stages of MTLE, which was further corroborated by low levels of immediate early gene expression in immature neurons. This situation was different when looking at immature astroglia, which were present in all MTLE cases, but their location and activity was very much associated with epileptiform activity. Specifically, their results indicated that immature astrocytes can migrate towards neuronal activity, e.g., the hilus, or directly contribute to generating seizures. Their results suggest an intriguing hypothesis that immature astroglia play a role as 'lossy capacitor' in initiating coordinated epileptiform activity. Astrocytes would usually prevent excessive neurotransmission and limit neuronal hyperactivity by soaking calcium intracellularly and then activate inhibitory GABAergic interneurons [15, 32]. Once immature astroglia are unable to maintain extracellular ionic homeostasis, functional alterations in their activity lead to aberrant modulation of the network. As mentioned in the chapter above, these results offer a new avenue in rather targeting reactive astroglia, e.g., immature astroglia as described by Ammothumkandy or those described above as reactive astroglia with an innate inflammatory response. It needs to be shown, however, if these astroglial populations are similar to each other or present a new cell type specific to the disease condition. Topic 10: Autoimmune encephalitis and NORSE In my final chapter, I would like to highlight a paper addressing the impact of autoimmunity to the pathogenesis of TLE. TLE is one of the syndromes linked to autoantibodies, such as anti-N-Methyl-D-aspartate (NMDA), anti-leucine-rich glioma-inactivated protein 1 (Gli1), or glutamic acid decarboxylase (GAD). Tröscher and coworker specifically addressed the latter syndrome of GAD-TLE, in which the immunopathogenesis remains enigmatic [41]. They reviewed imaging, serum and cerebral spinal fluid analysis and brain tissue in a consecutive series of 15 patients with GAD-TLE, including immunohistochemistry, multiplex fluorescent microscopy and transcriptomic analysis for inflammatory mediators and neuronal degeneration. In nine of their patients, they observed medio-temporal swelling and T2 signal increase within the first 6 years after the onset of symptoms, and all developed unilateral or bilateral hippocampal sclerosis. Early CSF analysis < 6 years after onset revealed intrathecal synthesis of IgGs. There were also high numbers of plasma cells in surgical brain tissue but no evidence for complement-mediated tissue damage. In contrast, there was a dense infiltration of CD8+ cytotoxic T cells, some of which had a specific resident memory T cell phenotype. Cytotoxic granzyme B-positive T cells attached to neurons were also recognized at the microscopic level. Their data convincingly showed that patients with GAD-TLE go through an early encephalitic stage (< 6 years) with CD8+ cytotoxic T lymphocytes mediated, antigen-driven neuronal cell loss but without signs of complement-mediated cell death. Subsequently, patients enter an apparently immunological inactive or low-active stage with ongoing seizures, probably caused by the structural damage to the temporal lobe. The observation of such an early stage of tissue damage is clinically most helpful to understand why immunotherapies usually do not lead to seizure freedom. A review from Bien and Bauer further highlighted what neuropathology teaches us about autoimmune encephalitis, e.g., NMDA and Gli1, autoimmune associated epilepsies including Rasmussen, paraneoplastic or GAD-TLE [8]. The authors argue in favor of a research-driven neuropathology approach to better understand these diseases. It is so important to receive morphological as well as temporal information over disease periods, and which can best be acquired by studying the affected brain tissue. Molecular technique will then broaden and support these data. Last but not least is a comprehensive neuropathology description of New-Onset Refractory Status Epilepticus (NORSE), including its subtype with a preceding febrile illness also known as FIRES (Febrile Infection-Related Epilepsy Syndrome). These conditions represent a most severe emergency condition in epileptology, e.g. status epilepticus (SE). Indeed, the prognosis of NORSE has a high rate of mortality. As a matter of fact, most cases of NORSE remain inexplicable despite extensive clinical evaluations, EEG studies, neuroimaging and laboratory testing. As mentioned above, neuropathology evaluations conducted in biopsies or autopsy tissue are helpful, therefore, to better identify an etiology in NORSE or FIRES. Hanin and coworkers collected published data from 64 cases of NORSE and FIRES, including 66 neuropathology tissue samples: 37 biopsies, 18 autopsies, 7 epilepsy surgery samples, 4 not otherwise specified [20]. In general, SE associated neuropathology findings include neuronal cell loss in the hippocampus, cerebellum and thalamus associated with reactive gliosis and often also microglial activation. Same findings can be observed in NORSE and FIRES, with the addition of perivascular T-cell infiltration in a number of cases. These lymphocytic infiltrates are similar to those described above, mainly being cytotoxic T cells. They have been confirmed by the neuropathological examination in 16 % of published cases and helped to solve the etiology. These histopathology findings also triggered a different therapeutic approach, including a high dose pulse of steroids or even tacrolimus, although this treatment had not resolved the symptoms. Meanwhile, autoimmune encephalitis has been identified as most frequent cause of NORSE and FIRES, and which call for more studies to better identify the molecular target for a successful therapy. Thus, this series of recent papers highlighted the ongoing research interest and which hopefully will help to develop rational therapeutic options in the near future. References 1. Aledo-Serrano A, Toledano R, Garcia-Morales I, Budke M, Beltran-Corbellini A, Blumcke I, Coras R, Gil-Nagel A (2022) D-galactose supplementation for the treatment of patients with mild malformation of cortical development with oligodendroglial hyperplasia in epilepsy (MOGHE): An interim analysis of a proof-of-concept trial. Epilepsia 63: 22-23 2. Ammothumkandy A, Ravina K, Wolseley V, Tartt AN, Yu PN, Corona L, Zhang N, Nune G, Kalayjian L, Mann JJ et al (2022) Altered adult neurogenesis and gliogenesis in patients with mesial temporal lobe epilepsy. Nat Neurosci 25: 493-503. https://doi.org/10.1038/s41593-022-01044-2 3. Aroor A, Nguyen P, Li Y, Das R, Lugo JN, Brewster AL (2023) Assessment of tau phosphorylation and beta-amyloid pathology in human drug-resistant epilepsy. Epilepsia Open 8: 609-622. https://doi.org/10.1002/epi4.12744 4. Atli B, Rath S, Burtscher J, Hainfellner JA, Hametner S (2022) Frontal intradiploic encephalocele in a 44-year-old male patient: illustrative case. J Neurosurg Case Lessons 4:. https://doi.org/10.3171/CASE2270 5. Baldassari S, Ribierre T, Marsan E, Adle-Biassette H, Ferrand-Sorbets S, Bulteau C, Dorison N, Fohlen M, Polivka M, Weckhuysen S et al (2019) Dissecting the genetic basis of focal cortical dysplasia: a large cohort study. Acta Neuropathol 138: 885-900. https://doi.org/10.1007/s00401-019-02061-5 6. Barba C, Blumcke I, Winawer MR, Hartlieb T, Kang HC, Grisotto L, Chipaux M, Bien CG, Hermanovska B, Porter BE et al (2023) Clinical Features, Neuropathology, and Surgical Outcome in Patients With Refractory Epilepsy and Brain Somatic Variants in the SLC35A2 Gene. Neurology 100: e528-e542. https://doi.org/10.1212/WNL.0000000000201471 7. Bedrosian TA, Miller KE, Grischow OE, Schieffer KM, LaHaye S, Yoon H, Miller AR, Navarro J, Westfall J, Leraas K et al (2022) Detection of brain somatic variation in epilepsy-associated developmental lesions. Epilepsia 63: 1981-1997. https://doi.org/10.1111/epi.17323 8. Bien CG, Bauer J (2023) What neuropathology teaches us about autoimmune encephalitides, autoimmune epilepsies, and encephalomyelitides. Clin Neuropathol 42: 87-92. https://doi.org/10.5414/NP301536 9. Blumcke I (2022) Neuropathology and epilepsy surgery: 2022 update. Free Neuropathology 3: 1-17. https://doi.org/doi.org/10.17879/freeneuropathology-2022-3813 10. Blumcke I, Coras R, Busch RM, Morita-Sherman M, Lal D, Prayson R, Cendes F, Lopes-Cendes I, Rogerio F, Almeida VS et al (2021) Toward a better definition of focal cortical dysplasia: An iterative histopathological and genetic agreement trial. Epilepsia 62: 1416-1428. https://doi.org/10.1111/epi.16899 11. Blumcke I, Thom M, Aronica E, Armstrong DD, Bartolomei F, Bernasconi A, Bernasconi N, Bien CG, Cendes F, Coras R et al (2013) International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: a Task Force report from the ILAE Commission on Diagnostic Methods. Epilepsia 54: 1315-1329. https://doi.org/10.1111/epi.12220 12. Bonduelle T, Hartlieb T, Baldassari S, Sim NS, Kim SH, Kang HC, Kobow K, Coras R, Chipaux M, Dorfmuller G et al (2021) Frequent SLC35A2 brain mosaicism in mild malformation of cortical development with oligodendroglial hyperplasia in epilepsy (MOGHE). Acta Neuropathol Commun 9: 3. https://doi.org/10.1186/s40478-020-01085-3 13. Chung C, Yang X, Bae T, Vong KI, Mittal S, Donkels C, Westley Phillips H, Li Z, Marsh APL, Breuss MW et al (2023) Comprehensive multi-omic profiling of somatic mutations in malformations of cortical development. Nat Genet 55: 209-220. https://doi.org/10.1038/s41588-022-01276-9 14. Concepcion FA, Ekstrom NA, Khan MN, Estes OO, Poolos NP (2023) Progressive Dysregulation of Tau Phosphorylation in an Animal Model of Temporal Lobe Epilepsy. Neuroscience 522: 42-56. https://doi.org/10.1016/j.neuroscience.2023.04.020 15. Deemyad T, Luthi J, Spruston N (2018) Astrocytes integrate and drive action potential firing in inhibitory subnetworks. Nat Commun 9: 4336. https://doi.org/10.1038/s41467-018-06338-3 16. Di Giacomo R, Burini A, Visani E, Doniselli FM, Cuccarini V, Garbelli R, Marucci G, De Santis D, Didato G, Deleo F et al (2023) Distinctive electro-clinical, neuroimaging and histopathological features of temporal encephaloceles associated to epilepsy. Neurol Sci 44: 4451-4463. https://doi.org/10.1007/s10072-023-06939-x 17. Eriksson MH, Ripart M, Piper RJ, Moeller F, Das KB, Eltze C, Cooray G, Booth J, Whitaker KJ, Chari A et al (2023) Predicting seizure outcome after epilepsy surgery: Do we need more complex models, larger samples, or better data? Epilepsia 64: 2014-2026. https://doi.org/10.1111/epi.17637 18. Garganis K, Gkiatis K, Maletic J, Harushukuri J, Kondylidis N, Dinopoulos A, Vorgia P, Coras R, Bluemcke I, Zountsas B (2023) Frontal lobe epilepsy and mild malformation with oligodendroglial hyperplasia: Further observations on electroclinical and imaging phenotypes, and surgical perspectives. Epileptic Disorders 25: 343-359. https://doi.org/10.1002/epd2.20062 19. Grote A, Heiland DH, Taube J, Helmstaedter C, Ravi VM, Will P, Hattingen E, Schure JR, Witt JA, Reimers A et al (2023) 'Hippocampal innate inflammatory gliosis only' in pharmacoresistant temporal lobe epilepsy. Brain 146: 549-560. https://doi.org/10.1093/brain/awac293 20. Hanin A, Cespedes J, Huttner A, Strelnikov D, Gopaul M, DiStasio M, Vezzani A, Hirsch LJ, Aronica E (2023) Neuropathology of New-Onset Refractory Status Epilepticus (NORSE). J Neurol 270: 3688-3702. https://doi.org/10.1007/s00415-023-11726-x 21. Hoffmann L, Coras R, Kobow K, López-Rivera JA, Lal D, Leu C, Najm I, Nürnberg P, Herms J, Harter PN et al (2023) Ganglioglioma with adverse clinical outcome and atypical histopathological features were defined by alterations in PTPN11 and other RAS-/MAP-Kinase pathway genes (vol 145, pg 815, 2023). Acta Neuropathologica 145: 851-855. https://doi.org/10.1007/s00401-023-02577-x 22. Holthausen H, Coras R, Tang Y, Bai L, Wang I, Pieper T, Kudernatsch M, Hartlieb T, Staudt M, Winkler P et al (2022) Multilobar unilateral hypoplasia with emphasis on the posterior quadrant and severe epilepsy in children with FCD ILAE Type 1A. Epilepsia 63: 42-60. https://doi.org/10.1111/epi.17114 23. Honke J, Hoffmann L, Coras R, Kobow K, Leu C, Pieper T, Hartlieb T, Bien CG, Woermann F, Cloppenborg T et al (2023) Deep histopathology genotype-phenotype analysis of focal cortical dysplasia type II differentiates between the GATOR1-altered autophagocytic subtype IIa and MTOR-altered migration deficient subtype IIb. Acta Neuropathol Com 11: ARTN 179. https://doi.org/10.1186/s40478-023-01675-x 24. Jirsa V, Wang H, Triebkorn P, Hashemi M, Jha J, Gonzalez-Martinez J, Guye M, Makhalova J, Bartolomei F (2023) Personalised virtual brain models in epilepsy. Lancet Neurol 22: 443-454. https://doi.org/10.1016/S1474-4422(23)00008-X 25. Kerr WT, McFarlane KN (2023) Machine Learning and Artificial Intelligence Applications to Epilepsy: a Review for the Practicing Epileptologist. Curr Neurol Neurosci Rep 23: 869-879. https://doi.org/10.1007/s11910-023-01318-7 26. Khoshkhoo S, Wang YL, Chahine Y, Erson-Omay EZ, Robert SM, Kiziltug E, Damisah EC, Nelson-Williams C, Zhu GY, Kong WN et al (2023) Contribution of Somatic Ras/Raf/Mitogen-Activated Protein Kinase Variants in the Hippocampus in Drug-Resistant Mesial Temporal Lobe Epilepsy. Jama Neurology 80: 578-587. https://doi.org/10.1001/jamaneurol.2023.0473 27. Kolble K, Cross JH, Becker A, Blumcke I (2018) A web-based diagnostic reference centre for the European Reference Network "EpiCare": recommendations of the eNeuropathology working group. Epileptic Disord 20: 339-345. https://doi.org/10.1684/epd.2018.1002 28. Lamberink HJ, Otte WM, Blümcke I, Braun KPJ (2020) Seizure outcome and use of antiepileptic drugs after epilepsy surgery according to histopathological diagnosis: a retrospective multicentre cohort study. Lancet Neurol 19: 748-757. https://doi.org/10.1016/s1474-4422(20)30220-9 29. Lerond J, Mathon B, Scopin M, Nichelli L, Guegan J, Bertholle C, Izac B, Andrieu M, Gareau T, Donneger F et al (2023) Hippocampal and neocortical BRAF mutant non-expansive lesions in focal epilepsies. Neuropathol Appl Neurobiol 49: e12937. https://doi.org/10.1111/nan.12937 30. Lopez-Rivera JA, Leu C, Macnee M, Khoury J, Hoffmann L, Coras R, Kobow K, Bhattarai N, Perez-Palma E, Hamer H et al (2022) The genomic landscape across 474 surgically accessible epileptogenic human brain lesions. Brain:. https://doi.org/10.1093/brain/awac376 31. Macdonald-Laurs E, Warren AEL, Francis P, Mandelstam SA, Lee WS, Coleman M, Stephenson SEM, Barton S, D'Arcy C, Lockhart PJ et al (2023) The clinical, imaging, pathological and genetic landscape of bottom-of-sulcus dysplasia. Brain:. https://doi.org/10.1093/brain/awad379 32. Mu Y, Bennett DV, Rubinov M, Narayan S, Yang CT, Tanimoto M, Mensh BD, Looger LL, Ahrens MB (2019) Glia Accumulate Evidence that Actions Are Futile and Suppress Unsuccessful Behavior. Cell 178: 27-43 e19. https://doi.org/10.1016/j.cell.2019.05.050 33. Najm I, Lal D, Vanegas MA, Cendes F, Lopes-Cendes I, Palmini A, Paglioli E, Sarnat HB, Walsh CA, Wiebe S et al (2022) The ILAE consensus classification of focal cortical dysplasia: An update proposed by an ad hoc task force of the ILAE diagnostic methods commission. Epilepsia 63: 1899-1919. https://doi.org/10.1111/epi.17301 34. Pitsch J, Kuehn JC, Gnatkovsky V, Muller JA, van Loo KMJ, de Curtis M, Vatter H, Schoch S, Elger CE, Becker AJ (2019) Anti-epileptogenic and Anti-convulsive Effects of Fingolimod in Experimental Temporal Lobe Epilepsy. Mol Neurobiol 56: 1825-1840. https://doi.org/10.1007/s12035-018-1181-y 35. Reinhardt A, Pfister K, Schrimpf D, Stichel D, Sahm F, al. e (2022) Anaplastic ganglioglioma – a diagnosis comprising several distinct tumour types. Neuropathology and Applied Neurobiology 48 (7): e12847. https://doi.org/10.1111/nan.12847 36. Ribierre T, Deleuze C, Bacq A, Baldassari S, Marsan E, Chipaux M, Muraca G, Roussel D, Navarro V, Leguern E et al (2018) Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J Clin Invest 128: 2452-2458. https://doi.org/10.1172/JCI99384 37. Schurr J, Coras R, Rossler K, Pieper T, Kudernatsch M, Holthausen H, Winkler P, Woermann F, Bien CG, Polster T et al (2017) Mild Malformation of Cortical Development with Oligodendroglial Hyperplasia in Frontal Lobe Epilepsy: A New Clinico-Pathological Entity. Brain Pathol 27: 26-35. https://doi.org/10.1111/bpa.12347 38. Sim NS, Seo Y, Lim JS, Kim WK, Son H, Kim HD, Kim S, An HJ, Kang HC, Kim SH et al (2018) Brain somatic mutations in SLC35A2 cause intractable epilepsy with aberrant N-glycosylation. Neurol Genet 4: e294. https://doi.org/10.1212/NXG.0000000000000294 39. Stone TJ, Keeley A, Virasami A, Harkness W, Tisdall M, Izquierdo Delgado E, Gutteridge A, Brooks T, Kristiansen M, Chalker J et al (2018) Comprehensive molecular characterisation of epilepsy-associated glioneuronal tumours. Acta Neuropathol 135: 115-129. https://doi.org/10.1007/s00401-017-1773-z 40. Stone TJ, Mankad K, Tan AP, Jan W, Pickles JC, Gogou M, Chalker J, Slodkowska I, Pang E, Kristiansen M et al (2023) DNA methylation-based classification of glioneuronal tumours synergises with histology and radiology to refine accurate molecular stratification. Neuropathol Appl Neurobiol 49: e12894. https://doi.org/10.1111/nan.12894 41. Tröscher AR, Mair KM, de Juan LV, Köck U, Steinmaurer A, Baier H, Becker A, Blümcke I, Finzel M, Geis C et al (2023) Temporal lobe epilepsy with GAD antibodies: neurons killed by T cells not by complement membrane attack complex. Brain 146: 1436-1452. https://doi.org/10.1093/brain/awac404 42. Vermeulen C, Pages-Gallego M, Kester L, Kranendonk MEG, Wesseling P, Verburg N, de Witt Hamer P, Kooi EJ, Dankmeijer L, van der Lugt J et al (2023) Ultra-fast deep-learned CNS tumour classification during surgery. Nature 622: 842-849. https://doi.org/10.1038/s41586-023-06615-2 43. Vorndran J, Neuner C, Coras R, Hoffmann L, Geffers S, Honke J, Herms J, Roeber S, Hamer H, Brandner S et al (2023) A deep learning-based histopathology classifier for Focal Cortical Dysplasia. Neural Comput Appl 35: 12775-12792. https://doi.org/10.1007/s00521-023-08364-9 44. Walger L, Adler S, Wagstyl K, Henschel L, David B, Borger V, Hattingen E, Vatter H, Elger CE, Baldeweg T et al (2023) Artificial intelligence for the detection of focal cortical dysplasia: Challenges in translating algorithms into clinical practice. Epilepsia 64: 1093-1112. https://doi.org/10.1111/epi.17522 45. Winawer MR, Griffin NG, Samanamud J, Baugh EH, Rathakrishnan D, Ramalingam S, Zagzag D, Schevon CA, Dugan P, Hegde M et al (2018) Somatic SLC35A2 variants in the brain are associated with intractable neocortical epilepsy. Ann Neurol 83: 1133-1146. https://doi.org/10.1002/ana.25243 46. Witt JA, Becker AJ, Helmstaedter C (2023) The multifactorial etiology of cognitive deficits in epilepsy and the neuropathology of mesial temporal lobe epilepsy beyond hyperphosphorylated tau. Alzheimers Dement 19: 3231-3232. https://doi.org/10.1002/alz.13085 47. Witters P, Tahata S, Barone R, Ounap K, Salvarinova R, Gronborg S, Hoganson G, Scaglia F, Lewis AM, Mori M et al (2020) Clinical and biochemical improvement with galactose supplementation in SLC35A2-CDG. Genet Med 22: 1102-1107. https://doi.org/10.1038/s41436-020-0767-8 48. Xi ZQ, Wang XF, Shu XF, Chen GJ, Xiao F, Sun JJ, Zhu X (2011) Is intractable epilepsy a tauopathy? Med Hypotheses 76: 897-900. https://doi.org/10.1016/j.mehy.2011.03.003 49. Zawar I, Kapur J (2023) Does Alzheimer's disease with mesial temporal lobe epilepsy represent a distinct disease subtype? Alzheimers Dement 19: 2697-2706. https://doi.org/10.1002/alz.12943
Copyright: © 2024 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |