|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Free Neuropathology 4:22 (2023) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Original Paper |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Molecular fragment characteristics and distribution of tangle associated TDP-43 (TATs) and other TDP-43 lesions in Alzheimer’s disease |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Keith A. Josephs1, Shunsuke Koga2, Nirubol Tosakulwong3, Stephen D Weigand3, Nha Trang Thu Pham4, Matt Baker2, Jennifer L. Whitwell4, Rosa Rademakers2,5, Leonard Petrucelli2, Dennis W. Dickson2 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Corresponding author: |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Submitted: 15 November 2023 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Keywords: TAR DNA binding protein 43, TMEM106b, Cluster analysis, TAT, Hippocampal sclerosis |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Abstract TAR DNA binding protein 43 (TDP-43) pathology is a defining feature of frontotemporal lobar degeneration (FTLD). In FTLD-TDP there is a moderate-to-high burden of morphologically distinctive TDP-43 immunoreactive inclusions distributed throughout the brain. In Alzheimer’s disease (AD), similar TDP-43 immunoreactive inclusions are observed. In AD, however, there is a unique phenomenon of neurofibrillary tangle-associated TDP-43 (TATs) whereby TDP-43 intermingles with neurofibrillary tangles. Little is known about the characteristics and distribution of TATs, or how burden and distribution of TATs compares to burden and distribution of other FTLD-TDP-like lesions observed in AD. Here we characterize molecular fragment characteristics, burden and distribution of TATs and assess how these features compare to features of other TDP-43 lesions. We performed TDP-43 immunohistochemistry with anti-phosphorylated, C- and N-terminal TDP-43 antibodies in 20 high-probability AD cases and semi-quantitative burden of seven inclusion types within five brain regions (entorhinal cortex, subiculum, CA1 and dentate gyrus of hippocampus, occipitotemporal cortex). Hierarchical cluster analysis was used to analyze the dataset that consisted of 75 different combinations of neuropathological features. TATs were nonspherical with heterogeneous staining patterns and present in all regions except hippocampal dentate. All three antibodies detected TATs although N-terminal antibody sensitivity was low. Three clusters were identified: Cluster-1 had mild-moderate TATs, moderate-frequent neuronal cytoplasmic inclusions, dystrophic neurites, neuronal intranuclear inclusions and fine neurites, and perivascular and granular inclusions identified only with the N-terminal antibody throughout the brain; Cluster-2 had scant TATs in limbic regions and Cluster-3 mild-moderate TATs and mild-moderate neuronal cytoplasmic inclusions and dystrophic neurites throughout the brain and moderate fine neurites. Only 17% of cluster 1 cases had the TMEM106b GG (protective) haplotype and 83% had hippocampal sclerosis. Both features differed across clusters (p=0.03 & p=0.01). TATs have molecular characteristics, distribution and burden, and genetic and pathologic associations like FTLD-TDP lesions. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Abbreviations AD - Alzheimer’s disease, DN - dystrophic neurites, ERC - entorhinal cortex, FTLD - frontotemporal lobar degeneration, FN - fine neurites, GN - granular inclusions, HCA - hierarchical cluster analysis, HpScl - hippocampal sclerosis, NCI - neuronal cytoplasmic inclusions, NFT - neurofibrillary tangle, MAPT - microtubular associated protein tau, NII - neuronal intranuclear inclusions, TAT - tangle associated TDP-43, TDP-43 - TAR DNA binding protein of 43 kDa, TMEM - transmembrane protein, PART - primary age-related tauopathy, PV - perivascular inclusions, OTC - occipitotemporal cortex, cTDP-43 - C-terminal fragment of TDP-43, nTDP-43 - N-terminal fragment of TDP-43, pTDP - phosphorylated TDP Introduction TAR DNA binding protein 43 (TDP-43) is a pathological protein strongly linked with frontotemporal lobar degeneration (FTLD) (1, 2). In FTLD-TDP there are widespread, characteristic histopathological lesions, including neuronal cytoplasmic inclusions (NCIs), dystrophic neurites (DNs), fine neurites (FNs) of the hippocampus, granular (sometimes referred to as small discrete NCIs)/globular inclusions (GNs), perivascular inclusions (PVs) and neuronal intranuclear inclusions (NIIs) (3-7). Each lesion type in FTLD-TDP has a signature molecular pattern of phosphorylated TDP-43 defined by relative amounts of C-terminal fragments to full-length TDP-43 (8-11). Since the discovery of TDP-43 in FTLD (1, 2), we and others have also found TDP-43 immunoreactive inclusions in the brains of patients with Alzheimer’s disease (AD) (12-16). In AD, as in FTLD-TDP, there are NCIs, DNs, FNs of the hippocampus, GNs, PVs and NIIs. In AD, however, we have identified a distinct inclusion whereby TDP-43 in the amygdala is found intermingled with the neurofibrillary tangle (NFT) (12, 17). Subsequently, others have confirmed our finding (18). This NFT-associated TDP-43 (TATs) is found in over 55% of AD cases with TDP-43, and hence could play an important role in AD pathogenesis. At present, little is known about TATs. It is unknown whether TATs are only deposited in the amygdala or are more widely distributed, whether the burden of TATs differs across different brain regions, and how the distribution and burden of TATs in different brain regions compare to the distribution and burden of other FTLD-TDP lesions found in AD. Lastly, the molecular characteristics of TATs is unknown, nor how its molecular characteristics compare to the molecular characteristics of other FTLD-TDP inclusions found in AD. In this study we aimed to assess the molecular characteristics, distribution and burden of TATs and compare these features to those of the other FTLD-TDP inclusions found in AD. We hypothesize that TATs will show a pattern of widespread distribution given the distribution of NFT in AD, and that TATs will show similar molecular fragment characteristics to NCIs. Material & Methods Case selection The database of the brain bank at Mayo Clinic, Jacksonville, Florida was queried to identify 20 consecutive cases, between January 2018 and July 2021, that received a neuropathologic diagnosis of high probability AD with TATs in the amygdala. A diagnosis of high probability AD was based on the National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of AD (19). High probability AD was selected given the widespread distribution of NFTs in these cases to ensure that we would capture the true distribution of TATs in AD. To be included in the study, all cases had to also have available paraffin blocks for additional molecular pathological analyses. Demographic characteristics of the 20 cases in the cohort are shown in Table 1. All but two cases (90%) were Caucasian and nine (45%) were male. The median age at death in the cohort was 82 years (range: 76-100 years) and median disease duration was 11 years (range: 5-19 years). Fifty percent of the cases had a family history of a neurodegenerative disease. All but one case (95%) had a final clinical diagnosis of an Alzheimer’s spectrum dementia diagnosis; one case (case 5) had a frontotemporal dementia diagnosis. Neuropathologic methods All cases had undergone neuropathologic assessment by a single expert neuropathologist (DWD) and had standardized tissue sampling and semi-quantitation of AD pathology. Thioflavin-S fluorescent microscopy was used for the evaluation of the distribution of senile plaques and NFTs which were then used to determine the Thal β-amyloid phase (20, 21) and the Braak NFT stage (22), respectively. Immunohistochemistry was performed on all 20 cases with an antibody to alpha-synuclein (NACP, 1:3000 rabbit polyclonal, Mayo Clinic antibody (23)) and with a phosphorylated tau antibody (PHF-1, 1:1000 mouse monoclonal, gift from Dr. Peter Davies). Hippocampal sclerosis (HpScl) was assessed on hematoxylin-eosin stained sections and was diagnosed based on the presence of neuronal loss and gliosis occurring in the subiculum and/or CA1 sector of the hippocampus (24). The cohort had a median Thal phase (21) of 5 (range: 4-5) and a median Braak NFT stage (22) of VI (range: V-VI) (Table 1). Eleven cases (55%) had amygdala-only Lewy bodies (25) and eight cases (40%) had HpScl (24). Table 1: Demographic and clinical features of the 20 cases
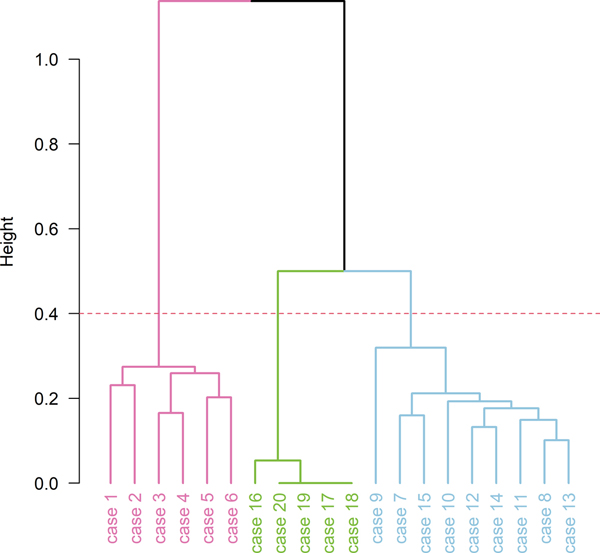
CAA = cerebral amyloid angiopathy; F = female; HpScl = hippocampal sclerosis; M = male; PI = Pacific Islander; PPA = primary progressive aphasia ¥ Suggestive of frontotemporal dementia; disease duration and age at death are shown in years. Ω Staging was determined prior to the study and was based only on pTDP-43 immunohistochemistry. TDP-43 immunohistochemistry & semi-quantitation For all 20 cases we performed serial sectioning and TDP-43 immunohistochemistry with three different TDP-43 antibodies: a commercially available phosphorylated TDP-43 (pTDP-43) antibody (pS409/410, 1:5000 mouse monoclonal, Cosmo Bio Co., LTD) that recognizes TDP-43 with phosphorylated epitopes; anti TDP-43 antibody that recognizes a neoepitope in the C terminal fragment of cleaved TDP-43(cTDP-43) (MC2085, 1:2500 rabbit polyclonal, gift from Leonard Petrucelli, Mayo Clinic) (8); and anti TDP-43 antibody that recognizes an epitope in the amino terminus (nTDP-43) (MC1079; 1:2500 rabbit polyclonal, gift from Leonard Petrucelli, Mayo Clinic) (8). Antigens for both non-commercial antibodies, as well as detailed biochemistry regarding both antibodies have been previously published (8, 26-28). Antibodies targeting phosphoserine 409 and 410 have been shown to have very strong immunoreactivity to TDP-43 inclusions in FTLD-TDP (9, 29). N-terminal TDP-43 fragments are believed to be rapidly degraded (30, 31) and as expected MC1079 binding represents detection of full length TDP-43(8). In all cases, immunohistochemistry was performed using a DAKO Autostainer (Universal Staining System, Carpinteria, CA). For this study, two investigators (KAJ & DWD) reviewed all 20 cases together and performed regional semi-quantitation of TDP-43 pathology with the three antibodies (pTDP-43, cTDP-43 & nTDP-43) in five regions: 1) entorhinal cortex (ERC); 2) CA1 sector of the hippocampus; 3) subiculum of the hippocampus; 4) dentate gyrus of the hippocampus and 5) occipitotemporal cortex (OTC). Lesions were considered positive, independent of the intensity of lesion staining. Hence, lesions showing slight/mild staining with one antibody were given equal weight to being positive to lesions with robust intensity with another antibody. That is, both staining patterns were assumed positive. This accounts for differences in affinities of the three different antibodies. For each region, TDP-43 deposition was assessed independently for seven different types of lesions: (1) small discrete NCIs; (2) short/comma-like DNs; (3) FNs in the CA1 region of the hippocampus as described by Hatanpaa et al (4); (4) PVs (typically bilobular and located next to capillary basal lamina) described by Lin et al (7); (5) TATs described by Amador-Ortiz(12); (6) grain- and globular-like intraparenchymal inclusions (GNs) and (7) NIIs. The burden of all lesions except for NIIs were scored semi-quantitatively based on a 5-point scale to be able to compare across lesion type: 0 = no inclusions identified, 0.5 = scant number of inclusions (1-3); 1= mild inclusions (4-10), 2= moderate inclusions (11-30) and 3 = marked/frequent number of inclusions (>30). For NIIs, we documented the presence/absence since when present, they were rare. For each lesion type we assessed the burden detected with all three anti-TDP-43 antibodies independently: pTDP-43; cTDP-43 and nTDP-43. Inclusion burden was assessed at a 200X magnification, with multiple visual fields of view, i.e., as many as necessary to examine the entire region of interest. Any possible NIIs that were observed at 200X magnification was confirmed as an NII at a 400X magnification. Hence, a total of 75 combinations of brain regions (n=5), inclusions (n=7) and antibodies (n=3) were analyzed (note: not every lesion was assessed across every region, e.g., FNs were only assessed in CA1). Confocal microscopy Immunofluorescence double-staining with the combinations of phospho-tau (CP13; mouse monoclonal; 1:1000; from the late Dr. Peter Davies, Feinstein Institute, North Shore Hospital, NY) and phospho-TDP-43 (Rb3655, rabbit polyclonal, 1:1000; Mayo Clinic antibody) was performed by SK who has expertise in the technique (32). The deparaffinized and rehydrated sections were steamed in distilled water for 30 minutes. Next, sections were blocked with Protein Block plus Serum Free (DAKO) for 1 hour and incubated with primary antibodies diluted in with Antibody Diluent with Background-Reducing Components (DAKO) overnight at 4°C. Sections were washed three times with 1xPBS at room temperature, and then incubated with secondary antibodies Alexa Fluor 568 and Alexa Fluor 488 (1:500, Thermo Fisher Scientific, Inc.) diluted with Antibody Diluent with Background-Reducing Components (DAKO) for 1.5 hours at room temperature in a dark chamber. Sections were washed three times with 1xPBS at room temperature, incubated with 1% Sudan Black for 2 minutes, washed with distilled water and mounted with Vectashield mounting media containing DAPI (Vector Laboratories). Representative images were taken with a confocal laser-scanning microscope (LSM 880; Carl Zeiss, Jena, Germany) using EC Plan-Neofluar 40x/1.30 Oil DIC M27 objective (Carl Zeiss). Genetic Screening Genomic DNA was extracted from the cerebellum of all twenty cases using the DNeasy blood and tissue kit (Qiagen Sciences) and genotyped using Taqman SNP assays analyzed with the QuantStudio 7 Flex Real-Time polymerase chain reaction (PCR) System (ThermoFisher). Genotypes were generated for APOE (rs429358 and rs7412) using Taqman assay ID C___3084793_20 and C____904973_10, GRN (rs5848) using C___7452046_20, MAPT (rs1052553) using C___7563736_10 and TMEM106B (rs3173615) using C__27465458_10. The rs1052553 polymorphism defines the MAPT H1/H2 haplotype. The rs3173615 variant in TMEM106B encodes the amino acid coding change Threonine to Serine at position 185, which been shown to be a risk factor for FTLD-TDP (39), and reduced TMEM106B homozygote carrier status has been found to be associated with TDP-43 in AD (33). Statistical methods Hierarchical Clustering analysis (HCA) was performed using a total of 75 features (combinations of brain regions/inclusions/antibodies). Burden of NIIs (12 features) were treated as binary variables, while the rest (63 features) were normalized into the range of 0 and 1. To perform HCA, a distance between 2 data points was defined using "Gower" (which is appropriate for a data set with different data types (numeric, binary, etc.)), with "Ward's" (variance was minimized within clusters) as a cluster method. HCA results are shown with a dendrogram; a tree-like diagram that shows the hierarchical relationship between all data points. Height (y axis) represents the distance between two clusters that contain those data points (the higher the height, the less similar those data points are). We assessed clustering tendency of the case by visualizing a plot of the first two principal components of the data set, and used the Hopkins statistics as a measure of spatial randomness. The Hopkins statistic was 0.66 which shows that the data was likely to have meaningful clusters. We used the Elbow, Silhouette, and Gap statistic to determine the optimal number of clusters. Bootstrap resampling along with the Jaccard similarity index were used to assess cluster stability. Jaccard similarity index values close to 1 indicate stable clusters, values < 0.6 indicate cluster is unstable. Demographic, genetic, and pathological characteristics were compared across clusters using Kruskal-Wallis Rank Sum test for continuous variables, and Fisher's Exact test for categorical variables. Data shown are median (IQR) or n (%). Statistical analyses were performed using R statistical software. Results Characteristics of TATs TATs were characterized by being nonspherical and slightly larger than NCIs with a heterogeneous staining pattern whereby, within the inclusion, some areas show darker staining than others (Figure 1). Confocal microscopy confirmed that TATs represent TDP-43 colocalized with paired helical filament tau (Figure 2). Tangle-associated TDP-43 was identified in all regions, except for in the dentate granular cells of the hippocampus, and was identified with all three antibodies, although sensitivity of detection with the nTDP-43 antibody that detects full length TDP-43 was low compared to the cTDP-43 and pTDP-43 antibodies (Figure 3). Burden findings across the cohort A summary of the lesion burden data by subject, region, inclusion type and antibody type are shown in Figure 3. There were differences identified across the three antibodies. The cTDP-43 and pTDP-43 antibodies both similarly detected NCIs, DNs, TATs, NIIs and FNs, although the cTDP-43 antibody was slightly less sensitive to burden (Figure 4). The nTDP-43 antibody, while less sensitive at detecting NCIs, DNs, TATs, NIIs and FNs compared to the other two antibodies (Figure 4), was more sensitive at detecting GNs and PVs (Figure 5). There were some differences identified across the five regions that are worth highlighting (Figure 3). First, the subiculum and CA1 were less likely to have NCIs, and, when present, tended to have lower burden than the ERC, dentate gyrus of the hippocampus and OTC. Of the four regions where TATs were found (ERC, CA1, subiculum and OTC), TATs were most common in ERC and subiculum, followed by CA1 and lastly the OTC. NIIs were only observed in ERC and OTC. GNs, when present, were least common in the OTC, while PVs, when present, where least common in the subiculum. TATs were more common than NCIs in the subiculum across all three antibodies but were less common than NCIs in the OTC, especially with the cTDP-43 antibody. TATs were not identified in the dentate while NCIs were common in this region. Cluster analysis findings The elbow, silhouette and gap statistic suggested optimal number of clusters of 2 to 4. However, the Jaccard similarity index for 3 clusters had the highest value (0.70), and the least instability (0.27), and hence we assessed 3 clusters in our data. The hierarchical cluster analysis dendrogram is shown in Figure 6. Cluster-1 consisted of six subjects, cluster 2 of five subjects and cluster 3 of nine subjects. Cluster-1 was characterized by moderate-frequent burden of NCIs and DNs across regions, high burden of FNs in CA1, mild PV and GN identified only with the nTDP-43 antibody and mild-moderate TATs throughout the brain; Cluster 2 had scant TATs only; and Cluster 3 mild-moderate burden of TATs and mild-moderate NCIs, DNs and FNs and scant-mild PV and GN identified predominantly with the nTDP-43 antibody (Figure 7). NIIs were found in 83% of Cluster 1 cases, 0% of Cluster 2 cases and 33% in Cluster 3 cases. Table 2 shows the demographic, genetic and pathological features across the three clusters. No statistically significant differences were observed in sex, family history, disease duration, age at death or APOE genotype. There was a significant difference in TMEM106B genotype for the rs3173615 SNP, with 17% of Cluster 1 cases have the protective (GG) allele, while 40% of Cluster 2 and 33% of Cluster 3 cases had the protective allele. There was also a significant difference in HpScl with the highest frequency observed in cluster 1 (83%) and the lowest frequency in cluster 2 (0%).
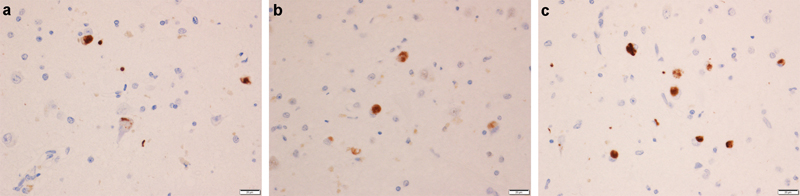
Figure 1: Morphological characteristics of tangle associated TDP-43 (TATs). Lesions are shown with anti-pTDP-43 antibody given that this antibody showed the most robust staining of TATs with different morphological characteristics seen in panels a, b, and c. Arrows point to TATs with different morphological characteristics.
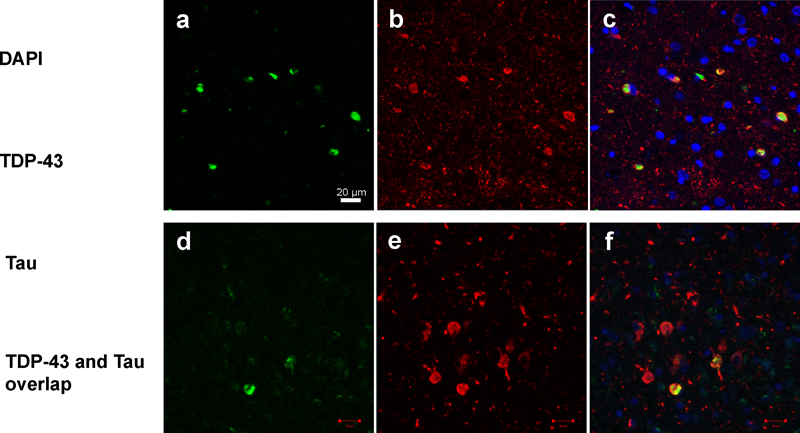
Figure 2: Confocal microscopy demonstrating overlap of phosphorylated TDP-43 and phosphorylated paired helical filament tau of TATs. Panels a and d show single labelling of TDP-43 immunoreactive inclusions. Panels b and e show single labelling of tau immunoreactive inclusions. Panels c and f show double labelling of TDP-43 and tau immunoreactive inclusions. Yellow color in panels c and f represents the overlap of the two proteins (TDP-43 and tau). DAPI (blue, single labelling) was used as a nuclear counterstain.
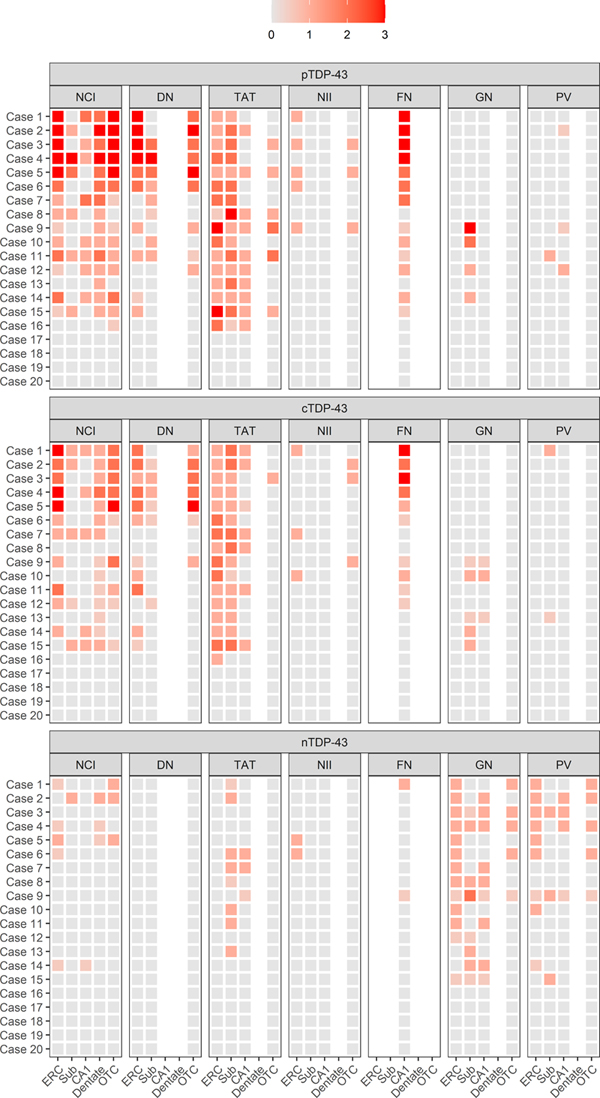
Figure 3: Distribution and burden of TDP immunoreactive lesions across regions for each case. The relationships between the three antibodies (pTDP-43, cTDP-43 and nTDP-43) and the seven lesion types (NCIs, DNs, FNs, NIIs, GNs, PVs, and TATs) across the five brain regions (entorhinal cortex (ERC), subiculum, CA1 of the hippocampus, dentate gyrus of the hippocampus and occipitotemporal cortex (OCT)) for each case. Brighter shades of orange represent higher burdens of pathology. Note NIIs were scored as present (1)/absent (0).
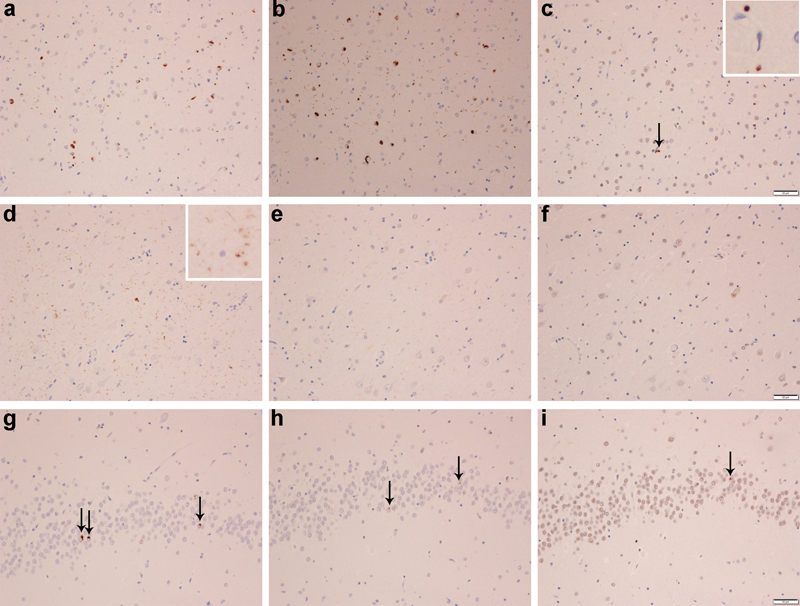
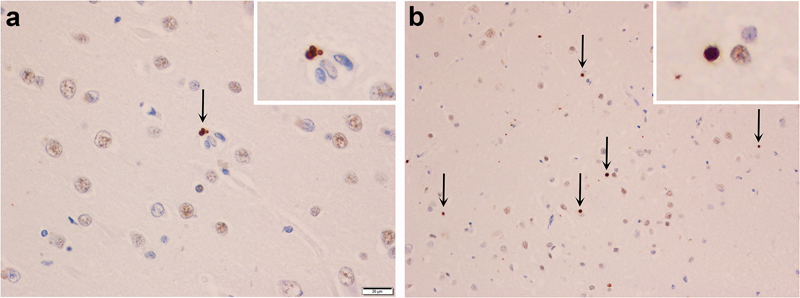
Figure 4: Phospho (pTDP), C-terminal fragment (cTDP) & full-length (nTDP) TDP-43 immunoreactivity of lesion types in AD. Labelling of neuronal cytoplasmic inclusions (NCIs), tangle associated TDP-43 (TATs) and dystrophic neurites (DNs) with pTDP-43 (a, d, g), cTDP-43 (b, e, h) and nTDP-43 (c, f, i) in the entorhinal cortex (Row 1), fine neurites (FN) in CA1 region of the hippocampus (Row 2) and NCIs in the dentate granule cells of the hippocampus (Row 3). The arrow in panel 4c points to an inclusion that is detected with nTDP-43 antibody, in the entorhinal cortex. The inset shows a magnified view of an inclusion labelled in the entorhinal cortex that has been labelled with the nTDP-43 antibody.
Figure 5: Full-length (nTDP) TDP-43 immunoreactivity of granular neurites and perivascular (PVs) inclusions. Granular neurites (a) and perivascular inclusions (b) in the occipitotemporal cortex (OTC) labelled with anti-nTDP-43 (full-length) antibodies.
Figure 6: Hierarchical cluster analysis dendrogram using all 75 features and highlighting the three clusters.
Figure 7: Distribution and burden of TDP immunoreactive lesions across regions for each cluster. The relationships between the three antibodies (pTDP-43, cTDP-43 and nTDP-43) and the seven lesion types (NCIs, DNs, FNs, NIIs, GNs, PVs, and TATs) across the five brain regions (entorhinal cortex (ERC), subiculum, CA1 of the hippocampus, dentate gyrus of the hippocampus and occipitotemporal cortex (OCT)) for each cluster. Brighter shades of orange represent higher average burdens of pathology within cluster. Note NIIs were scored as present (3)/absent (0) to put them on the same scale as the other inclusions. Table 2: Participants characteristics
Overall p-values for continuous variables are from Kruskal-Wallis Rank Sum test, and Fisher's Exact test for categorical variables. Data shown are median (IQ) or n (%) * Cluster 2 is statistically different from Cluster 3 (p = 0.009); † Cluster 1 is statistically different from Cluster 2 (p = 0.02); APOE = apolipoprotein E; MAPT = microtubule associated protein tau; GRN = progranulin; TMEM106B = transmembrane protein 106B; LBD = Lewy body disease Discussion In this study, we focus on TAT lesions that we described in the brains of patients with AD (17). We demonstrate that TATs are a mixed inclusion, characterized by the intermingling of two proteins: tau and TDP-43. We show that morphological characteristics of TATs are heterogeneous across lesions and that the burden of TATs vary by region within individual brains, as well as in the same region across different cases. We found that the molecular characteristics of TATs is identical to that of NCIs in AD. Lastly, we showed that TATs can be limited to being present in the amygdala only in AD although they are more often widespread and associated with the presence of NCIs, DNs, NIIs, FNs and other FTLD-TDP characteristic lesions; a relationship that appears to have genetic underpinnings. Neurofibrillary tangle-associated TDP-43 (TATs) have only been described in AD (12, 17, 18), unsurprisingly, given that NFTs are one of the two characterizing lesions in AD, the other being the senile plaque (19). TATs have not been described in FTLD-TDP, likely because NFTs tend to be absent, or no more than scant and limited in distribution when present in FTLD-TDP. With-that-said, we hypothesize that TATs should also be observed in other diseases in which NFTs are present, such as primary age-related tauopathy (PART) (34) since TDP-43 has been reported in PART (35-37). However, given the limited distribution and burden of NFTs in PART, one would not expect TATs to be as frequent as in AD, and certainly uncommonly found outside of limbic regions given that Braak and Braak stage is < IV in PART (34). We observed heterogeneous morphologies across TAT inclusions, often having the appearance of a bitten apple, or flame shaped. Under the light microscope, we also observed a heterogeneous staining pattern in individual lesions whereby different parts of the lesions had more and less robust (dark) staining. The reason for this is unclear but could be due to how much TDP-43 vs how much tau is being exposed on the surface that is being viewed. Another reason for the heterogeneous appearance of the inclusion may be due to the relative amounts of the different molecular fragments that make up the TDP-43 inclusions. We found TATs to be most robustly stained by pTDP-43 antibodies suggesting that phosphorylated TDP-43 is the main composition of TATs. There was an almost equal staining observed with the cTDP-43 antibody suggesting that the main composition of TATs are C-terminal fragments of TDP-43. Interestingly, there was some staining, although much less robust, with the nTDP-43 antibody that recognizes full-length TDP-43. This suggests that TATs, while consisting of predominantly phosphorylated C-terminal fragments of TDP-43, also consist of some full-length TDP-43. One of the interesting findings of this study was that the molecular characteristics of TATs mirrored that of NCIs. NCIs were indeed smaller than TATs, but, like TATs, also showed the most robust staining with the pTDP-43 antibody followed closely by staining with the cTDP-43 antibody and much less staining occurring with the nTDP-43 antibody. We have previously found the identical molecular staining pattern in NCIs in FTLD-TDP (11). Hence, NCIs in FTLD-TDP, NCIs in AD and TATs share similar molecular characteristics. What was interesting was the fact that although all cases in this study were Braak stage V-VI with NFTs extending into association and primary neocortices, TATs were predominantly deposited in limbic regions (amygdala + ERC and hippocampal subregions) although not in the dentate gyrus of the hippocampus. The burden of TATs in the OCT, a non-limbic region, was much less compared to the amount observed in limbic regions. This was different from what we observed for NCIs, which in many instances showed an equal burden, or even higher burden in the OCT compared to limbic regions. There were 75 different combinations of antibodies, lesions and regions which made it difficult to visually determine whether there were distinct relationships within the cohort. However, when we applied cluster analysis, we found three different clusters. The first (Cluster 1, N=6) was characterized by a minimal burden of TATs, as well as widespread deposition of FTLD-TDP inclusions. All cases in this cluster had FNs of the CA1 region of the hippocampus, and the majority (83%) were also found to have NIIs and HpScl. Hence, Cluster 1 has features that are highly reminiscent of FTLD-TDP (3, 5) and, therefore, could be best summarized as FTLD-TDP with TATs. Cluster 2 (n=5) was very different from Cluster 1. In four of the five cases in cluster 2 there were no inclusions whatsoever identified with any of the three antibodies. The presence of TDP-43 was limited to the amygdala in four of the cases, with the fifth case only having scant TATs in limbic cortex with a single NCI identified in OCT with the pTDP-43 antibody. This cluster, therefore, had no features suggestive of FTLD-TDP and could best be summarized as one with focal amygdala TATs. The third cluster was the largest (n=9) and consisted of a moderate-marked number of TATs in limbic regions and in OCT. There were also FTLD-TDP inclusions present throughout the brain, but they had a lower burden compared to cluster 1. Interestingly, NIIs and HpScl were also present, albeit in only 33% of the cases. Hence, cluster 3 could be best summarized as having widespread TATs with some features of FTLD-TDP. Therefore, overall, it appears that approximately 50% of this cohort of high probability AD cases, all of the cases in Cluster 1 and for argument’s sake half of the case of Cluster 3, have salient features of FTLD-TDP. We found evidence for an association between cluster and the TMEM106b alleles. TMEM106B has been found to be associated with an increased risk of having FTLD-TDP pathology (38-41). More specifically, the GG haplotype is associated with a decreased risk of FTLD-TDP (40). Only one of the five cases in Cluster 1 (FTLD-TDP with TATs) had the protective GG haplotype while 40% of the cases in Cluster 2 and 33% of the cases in Cluster 3 had the GG haplotype. Of note, approximately 20% of normal controls vs only 6% of FTLD-TDP subjects have the GG haplotype (40). Hence, the absence of the GG haplotype in Cluster 1 is another characteristic of FTLD-TDP. Although the pTDP-43 and cTDP-43 antibodies were the best at staining NCIs, DNs, NIIs, FNs of the CA1 region of the hippocampus, as we have previously shown in FTLD-TDP (11), and TATs, we found that PVs and GNs were best identified with the nTDP-43 antibody. This suggests that PVs and GNs have a different molecular composition from the other FTLD-TDP-like inclusions in AD. The fact that PVs and GNs were best observed with the nTDP-43 antibody that recognizes full-length TDP-43 suggests that PVs and GNs may not be pathologic. Another explanation could be that PVs and GNs represent early inclusions that were not yet cleaved by caspases into C terminal fragments (8). There are strengths and limitations to this study that are worth mentioning. In terms of strengths, our sample was unbiased as we selected consecutive cases for the study; pathological determination of Braak NFT stage was performed by a single experienced neuropathologist, reducing variability, and TDP-43 inclusion assessments were performed by two experts to reach consensus on severity ratings; and we performed confocal microscopy to confirm that TATs represent TDP-43 colocalized with paired helical filament tau. Limitations of the study include the lack of racial heterogeneity in our sample which may limit our findings to Caucasians, and the fact that we did not have volumetric head MRI or [18F] fluorodeoxyglucose PET scans to determine whether our clusters had different antemortem signatures of atrophy or hypometabolism. In conclusion, we have identified key characteristics of an understudied pathological lesion that is common in AD, that we refer to as TATs (tangle-associated TDP-43). It appears that the association between TATs and lesions characteristic of FTLD-TDP is not random but instead may have a genetic underpinning. Declarations Ethics approval and consent to participate The Mayo institutional Review Board (IRB) has deemed this study exempt from IRB review and approval given that it utilizes postmortem brain tissue only. Consent for publication Not applicable Availability of data and material The datasets generated and analyzed for the current study is available from the corresponding author on reasonable request. Competing interests None Funding The study was funded by National Institute of Aging grants R01-AG37491 and RF-NS120992. Authors' contributions
Dr. Josephs was responsive for generating TDP-43 burden data, obtaining study funding, and for drafting the manuscript. Acknowledgements We wish to thank Ms. Monica Casey Castanedes for pathological support. References 1. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130-3. DOI: 10.1126/science.1134108. 2. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602-11. DOI: 10.1016/j.bbrc.2006.10.093. 3. Josephs KA, Stroh A, Dugger B, Dickson DW. Evaluation of subcortical pathology and clinical correlations in FTLD-U subtypes. Acta Neuropathol. 2009;118(3):349-58. DOI: 10.1007/s00401-009-0547-7. 4. Hatanpaa KJ, Bigio EH, Cairns NJ, Womack KB, Weintraub S, Morris JC, et al. TAR DNA-binding protein 43 immunohistochemistry reveals extensive neuritic pathology in FTLD-U: a midwest-southwest consortium for FTLD study. J Neuropathol Exp Neurol. 2008;67(4):271-9. DOI: 10.1097/NEN.0b013e31816a12a6. 5. Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114(1):5-22. DOI: 10.1007/s00401-007-0237-2. 6. Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119(1):1-4. DOI: 10.1007/s00401-009-0612-2. 7. Lin WL, Castanedes-Casey M, Dickson DW. Transactivation response DNA-binding protein 43 microvasculopathy in frontotemporal degeneration and familial Lewy body disease. J Neuropathol Exp Neurol. 2009;68(11):1167-76. DOI: 10.1097/NEN.0b013e3181baacec. 8. Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD, et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(18):7607-12. DOI: 10.1073/pnas.0900688106. 9. Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol. 2008;64(1):60-70. DOI: 10.1002/ana.21425. 10. Igaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, Neumann M, et al. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. The American journal of pathology. 2008;173(1):182-94. DOI: 10.2353/ajpath.2008.080003. 11. Josephs KA, Zhang YJ, Baker M, Rademakers R, Petrucelli L, Dickson DW. C-terminal and full length TDP-43 specie differ according to FTLD-TDP lesion type but not genetic mutation. Acta Neuropathol Commun. 2019;7(1):100. DOI: 10.1186/s40478-019-0755-x. 12. Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol. 2007;61(5):435-45. DOI: 10.1002/ana.21154. 13. Arai T, Mackenzie IR, Hasegawa M, Nonoka T, Niizato K, Tsuchiya K, et al. Phosphorylated TDP-43 in Alzheimer's disease and dementia with Lewy bodies. Acta Neuropathol. 2009;117(2):125-36. DOI: 10.1007/s00401-008-0480-1. 14. Josephs KA, Whitwell JL, Knopman DS, Hu WT, Stroh DA, Baker M, et al. Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology. 2008;70(19 Pt 2):1850-7. DOI: 10.1212/01.wnl.0000304041.09418.b1. 15. Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM, et al. TDP-43 is a key player in the clinical features associated with Alzheimer's disease. Acta Neuropathol. 2014;127(6):811-24. DOI: 10.1007/s00401-014-1269-z. 16. Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain. 2019;142(6):1503-27. DOI: 10.1093/brain/awz099. 17. Josephs KA, Murray ME, Tosakulwong N, Weigand SD, Serie AM, Perkerson RB, et al. Pathological, imaging and genetic characteristics support the existence of distinct TDP-43 types in non-FTLD brains. Acta Neuropathol. 2019;137(2):227-38. DOI: 10.1007/s00401-018-1951-7. 18. Tome SO, Vandenberghe R, Ospitalieri S, Van Schoor E, Tousseyn T, Otto M, et al. Distinct molecular patterns of TDP-43 pathology in Alzheimer's disease: relationship with clinical phenotypes. Acta Neuropathol Commun. 2020;8(1):61. DOI: 10.1186/s40478-020-00934-5. 19. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123(1):1-11. DOI: 10.1007/s00401-011-0910-3. 20. Murray ME, Lowe VJ, Graff-Radford NR, Liesinger AM, Cannon A, Przybelski SA, et al. Clinicopathologic and 11C-Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer's disease spectrum. Brain. 2015;138(Pt 5):1370-81. DOI: 10.1093/brain/awv050. 21. Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58(12):1791-800. DOI: 10.1212/wnl.58.12.1791. 22. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl). 1991;82(4):239-59. DOI: 10.1007/BF00308809. 23. Gwinn-Hardy K, Mehta ND, Farrer M, Maraganore D, Muenter M, Yen SH, et al. Distinctive neuropathology revealed by alpha-synuclein antibodies in hereditary parkinsonism and dementia linked to chromosome 4p. Acta Neuropathol. 2000;99(6):663-72. DOI: 10.1007/s004010051177. 24. Dickson DW, Davies P, Bevona C, Van Hoeven KH, Factor SM, Grober E, et al. Hippocampal sclerosis: a common pathological feature of dementia in very old (> or = 80 years of age) humans. Acta Neuropathol. 1994;88(3):212-21. DOI: 10.1007/BF00293396. 25. Uchikado H, Lin WL, DeLucia MW, Dickson DW. Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol. 2006;65(7):685-97. DOI: 10.1097/01.jnen.0000225908.90052.07. 26. Carlomagno Y, Zhang Y, Davis M, Lin WL, Cook C, Dunmore J, et al. Casein kinase II induced polymerization of soluble TDP-43 into filaments is inhibited by heat shock proteins. PLoS One. 2014;9(3):e90452. DOI: 10.1371/journal.pone.0090452. 27. Chew J, Cook C, Gendron TF, Jansen-West K, Del Rosso G, Daughrity LM, et al. Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol Neurodegener. 2019;14(1):9. DOI: 10.1186/s13024-019-0310-z. 28. Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes-Casey M, et al. Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science. 2015;348(6239):1151-4. DOI: 10.1126/science.aaa9344. 29. Inukai Y, Nonaka T, Arai T, Yoshida M, Hashizume Y, Beach TG, et al. Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Lett. 2008;582(19):2899-904. DOI: 10.1016/j.febslet.2008.07.027. 30. Pesiridis GS, Tripathy K, Tanik S, Trojanowski JQ, Lee VM. A "two-hit" hypothesis for inclusion formation by carboxyl-terminal fragments of TDP-43 protein linked to RNA depletion and impaired microtubule-dependent transport. J Biol Chem. 2011;286(21):18845-55. DOI: 10.1074/jbc.M111.231118. 31. Buratti E. TDP-43 post-translational modifications in health and disease. Expert Opin Ther Targets. 2018;22(3):279-93. DOI: 10.1080/14728222.2018.1439923. 32. Koga S, Kouri N, Walton RL, Ebbert MTW, Josephs KA, Litvan I, et al. Corticobasal degeneration with TDP-43 pathology presenting with progressive supranuclear palsy syndrome: a distinct clinicopathologic subtype. Acta Neuropathol. 2018;136(3):389-404. DOI: 10.1007/s00401-018-1878-z. 33. Rutherford NJ, Carrasquillo MM, Li M, Bisceglio G, Menke J, Josephs KA, et al. TMEM106B risk variant is implicated in the pathologic presentation of Alzheimer disease. Neurology. 2012;79(7):717-8. DOI: 10.1212/WNL.0b013e318264e3ac. 34. Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128(6):755-66. DOI: 10.1007/s00401-014-1349-0. 35. Josephs KA, Murray ME, Tosakulwong N, Whitwell JL, Knopman DS, Machulda MM, et al. Tau aggregation influences cognition and hippocampal atrophy in the absence of beta-amyloid: a clinico-imaging-pathological study of primary age-related tauopathy (PART). Acta Neuropathol. 2017;133(5):705-15. DOI: 10.1007/s00401-017-1681-2. 36. Zhang X, Sun B, Wang X, Lu H, Shao F, Rozemuller AJM, et al. Phosphorylated TDP-43 Staging of Primary Age-Related Tauopathy. Neurosci Bull. 2019;35(2):183-92. DOI: 10.1007/s12264-018-0300-0. 37. Elobeid A, Libard S, Leino M, Popova SN, Alafuzoff I. Altered Proteins in the Aging Brain. J Neuropathol Exp Neurol. 2016;75(4):316-25. DOI: 10.1093/jnen/nlw002. 38. Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42(3):234-9. DOI: 10.1038/ng.536. 39. Pottier C, Zhou X, Perkerson RB, 3rd, Baker M, Jenkins GD, Serie DJ, et al. Potential genetic modifiers of disease risk and age at onset in patients with frontotemporal lobar degeneration and GRN mutations: a genome-wide association study. Lancet Neurol. 2018;17(6):548-58. DOI: 10.1016/S1474-4422(18)30126-1. 40. van Blitterswijk M, Mullen B, Nicholson AM, Bieniek KF, Heckman MG, Baker MC, et al. TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia. Acta Neuropathol. 2014;127(3):397-406. DOI: 10.1007/s00401-013-1240-4. 41. Gallagher MD, Suh E, Grossman M, Elman L, McCluskey L, Van Swieten JC, et al. TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol. 2014;127(3):407-18. DOI: 10.1007/s00401-013-1239-x.
Copyright: © 2023 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||