|
|
|
Free Neuropathology 5:4 (2024) |
|
Original Paper |
|
Blocking TGF-β- and Epithelial-to-Mesenchymal Transition (EMT)-mediated activation of vessel-associated mural cells in glioblastoma impacts tumor angiogenesis |
|
Luisa Merk1, Katja Regel1, Hermann Eckhardt1, Marietheres Evers1, Ali El-Ayoubi1, Michel Mittelbronn2-7, Marcel Krüger8, Jean-Jacques Gérardy3,7, Andreas F. Mack9, Ulrike Naumann1, 10 |
|
|
Corresponding author: |
|
Additional resources and electronic supplementary material: supplementary material |
|
Submitted: 10 November 2023 |
|
Keywords: Glioblastoma, Tumor vasculature, Pericytes, TGF-β, EMT |
|
Abstract Glioblastoma (GBM) is the most common malignant primary brain tumor in adults. GBM displays excessive and unfunctional vascularization which may, among others, be a reason for its devastating prognosis. Pericytes have been identified as the major component of the irregular vessel structure in GBM. In vitro data suggest an epithelial-to-mesenchymal transition (EMT)-like activation of glioma-associated pericytes, stimulated by GBM-secreted TGF-β, to be involved in the formation of a chaotic and dysfunctional tumor vasculature. This study investigated whether TGF-β impacts the function of vessel associated mural cells (VAMCs) in vivo via the induction of the EMT transcription factor SLUG and whether this is associated with the development of GBM-associated vascular abnormalities. Upon preventing the TGF-β-/SLUG-mediated EMT induction in VAMCs, the number of PDGFRβ and αSMA positive cells was significantly reduced, regardless of whether TGF-β secretion by GBM cells was blocked or whether SLUG was specifically knocked out in VAMCs. The reduced amount of PDGFRβ+ or αSMA+ cells observed under those conditions correlated with a lower vessel density and fewer vascular abnormalities. Our data provide evidence that the SLUG-mediated modulation of VAMC activity is induced by GBM-secreted TGF-β and that activated VAMCs are key contributors in neo-angiogenic processes. We suggest that a pathologically altered activation of GA-Peris in the tumor microenvironment is responsible for the unstructured tumor vasculature. There is emerging evidence that vessel normalization alleviates tumor hypoxia, reduces tumor-associated edema and improves drug delivery. Therefore, avoiding the generation of an unstructured and non-functional tumor vasculature during tumor recurrence might be a promising treatment approach for GBM and identifies pericytes as a potential novel therapeutic target. |
|
Introduction Glioblastoma (GBM) is the most common malignant primary brain tumor in adults with an incidence of 2 to 5 cases per 100,000 people in North America and Europe. GBM accounts for more than 50 % of primary malignant brain tumors. Worldwide, the number of new cases per year can be estimated at around 250,000. The median age at diagnosis is 64 years and it is more common in men as compared to women (for review see [1]). Its mean progression-free survival is just a few months. By standard therapy using optimal surgical resection, radiation and chemotherapy with temozolomide, or even using more recent additional treatment approaches such as tumor treating fields (TTF), there is invariably tumor recurrence. However, this combined treatment only slightly prolongs the median survival, but usually not more than up to 20 months [2]. Several characteristics of GBM account for its poor prognosis: (i) GBMs are highly invasively growing tumors which makes a complete surgical resection impossible. Even after aggressive surgery, tumor cells that invaded healthy brain areas will remain and are the source of recurrence [3]. (ii) GBMs show massive proliferation which makes them grow rapidly. The growing tumor mass is in turn the cause of several neurological problems such as seizures, impaired vision, drowsiness, changes in personality or massive headache, dependent on the location of the tumor in the brain [1]. (iii) GBMs can be categorized as highly immunosuppressive tumors. Several mechanisms such as the expression of immune checkpoint proteins like programmed cell death ligand 1 (PD-L1), the secretion of immunosuppressive cytokines such as transforming growth factor (TGF)-β by GBM cells or tumor vessel associated pericytes [4], the downregulation of major histocompatibility complex (MHC) class I molecules on GBM cells as well as metabolic changes in the tumor cells and the tumor microenvironment (TMA) negatively influence the immune surveillance of GBMs [5-8]. (vi) Glioma is a highly treatment resistant type of cancer [9]. (vii) Finally, GBMs are highly vascularized tumors, and the interaction between GBM cells and blood vessels promotes neoangiogenesis, thereby facilitating tumor growth [10]. Numerous mechanisms contribute to the robust formation of blood vessels within the tumor. This involves endothelial cell (EC) proliferation, which is dependent on hypoxia and hypoxia inducible factor (HIF)-1 induced transcription and release of vascular endothelial growth factor (VEGF) from GBM cells. This process leads to the sprouting of capillaries from pre-existing blood vessels. The release of further angiogenic factors from GBM cells recruits a variety of cells that may also participate in new blood vessel formation (for review see [11]). In GBM, the newly formed vessels often show abnormalities in shape, size and complexity, resulting in glomeruloid, garland-like or clustered bizarre vascular formations, which negatively influences the patient´s outcome [12]. However, not only ECs are involved in angiogenic processes and vessel formation. Especially pericytes came into focus to be the responsible cell type for the chaotic vessel structure in GBM, since these cells are heavily modified under pathological conditions (reviewed in [13, 14]). Pericytes are cells adjacent to ECs, wrapping around blood vessels. In the CNS, pericytes are necessary for the formation and regulation of the blood-brain-barrier (BBB). In former studies we have demonstrated that pericytes are prominently involved in the formation of GBM-associated vascular proliferations and that those pericytes covering GBM-associated vessels can be distinguished from "normal" pericytes covering vessels outside the tumor core by their expression profile, especially by the expression of epithelial-to-mesenchymal transition (EMT) factors such as SLUG or TWIST [15]. In vitro, GBM cells, by secretion of TGF-β, induce an EMT-like program in human brain microvascular pericytes, drive these cells to change their growth morphology, shift them towards energy production by glycolysis, induce proliferation and increase cell motility, and impact the BBB integrity [16, 17]. In this study, we were interested in whether the TGF-β-mediated induction of the EMT program in VAMCs might have impact on the density, formation and structure of newly built blood vessels in the tumor core of GBM, particularly in a murine mouse model in vivo. Material and Methods Cell culturing and testing Primary murine brain vascular pericytes (MBVP) were purchased from iXCells (San Diego, CA, USA) and were cultured in fully supplemented mouse pericyte growth medium (PM, iXCells). MBVPs were used up to passage 5. GL261 mouse glioma cells (Cellosaurus CVCL_Y003), known to express TGF-β [18], were from the Institute's repository. GL261 cells were created in 1970 via chemical induction with methylcholanthrene pellets implanted into the brains of C57BL/6 mice [19]. NIH/3T3 cells were from ATCC (#CRL-1658). GL261 and NIH/3T3 cells were cultivated in Dulbecco's Modified Eagles Medium (DMEM; Sigma-Aldrich, Taufkirchen, Germany) supplemented with 10% FBS, 100 U/mL penicillin, and 0.1 mg/mL streptomycin (Sigma-Aldrich) at 37 °C, 5 % CO2. All cell lines were regularly tested to be free of mycoplasma using the MycoAlert Mycoplasma Detection Kit (Lonza, Cologne, Germany). Generation and characterization of GL261 TGF-β1/β2 double knockout, mCherry expressing cells For the generation of GL261 TGF-β1 plus -β2 double knockout cells, the cells were transfected by electroporation with Alt-R Cas9 Nuclease, ATTO 550 CRISPR-Cas9 tracrRNA, mouse TGFB1 guide RNAs (GGCAGUAGCCGCAGCCCCGAGUUUUAGAGCUAUGCU and GUACAAAGCGAGCACCGCCUGUUUU-AGA-GCUAUGCU; all from Integrated DNA Technology, Coralville, IA, USA) using the Amaxa Cell Line Nucleofactor™ Kit V (Lonza) as described [20]. Guide RNAs were designed using Benchling (https://www.benchling.com). An in silico off-target binding prediction was performed using the online tool Off-Spotter (https://cm.jefferson.edu/Off-Spotter; [21]). After electroporation, transfected cells were sorted on a SH800 cell sorter (Sony, Stuttgart, Germany). After expansion of cell clones, biallelic TGF-β1 knockout GL261 cell clones were identified by PCR on genomic DNA on a PTC-200 PCR cycler (BioRad, Feldkirchen, Germany) using the following detection primer: mTGFB1-frw GGGTTCCCGCTCTCCGAAGTG and mTGFB1-rev GTCCACCATTAGCACGCGGG (Sigma). TGF-β1-KO cells were subsequently transfected using mouse TGFB2 guide RNAs (GCUCCGCUCGCGCUCGCAGGG-UUUUAGAGCUAUGCU and GCGCCACCGGGACCAGAUGCGUUUUAGAGCUAUGCU; Integrated DNA Technology) as described above. After sorting and clonal expansion as described above, the TGF-β2 knockout was analyzed by PCR on genomic DNAusing TGF-β2 specific primer (mTGFB2-frw CGTTTCTTCCTTTTAAAAACATG and mTGFB2-rev TGTCGATTTTATAAACCTCCTTG; all from Sigma). Further validation of the double knockout was performed by sequencing on an ABI Sequencer using BigDye® Terminator v 3.0 sequencing reaction mix (Applied Biosciences, Carlsbad, Germany). GL261 as well as GL261-TGF-β-KO cells were grown up and were transduced twice with Lenti-mCherry (Addgene; #36084). GL261mCherry (PAR) and GL261mCherry-TGF-β1/2-Knockout (TGF-β-KO) cells were subsequently characterized in terms of their proliferation and migration capacity. For this, 2.000 cells were seeded in microtiter plates and allowed to attach. 4 h after seeding and subsequently every 24 h later, cell density was determined by staining the cells with crystal violet as described [22]. To determine differences in cell motility, the transwell boyden chamber migration assay was used as described [23]. Briefly, 25.000 PAR or TGF-β-KO cells were treated for 3 h with mitomycin (0.5 µg/ml, Sigma) to block proliferation, were washed to remove residual mitomycin, then seeded in the top chamber of an 8 µm pore migration cassette (Corning, Kaiserslautern, Germany), and were allowed to migrate for 24 h. Migrated cells on the bottom side were stained with crystal violet and were counted manually. As attraction medium, conditioned medium from NIH/3T3 cells was used. Identification of the murine RGS5 minimal promoter DNA fragments of different lengths located in the mRGS5 promoter region (ensemble ID GRCm39:CM000994.3) were amplified by PCR from genomic DNA of C57BL6 using different primer combinations (F1 and F4, R1-R4; Suppl. Table 1, Suppl. Fig. 1). NheI and XbaI recognition sites were added to the 5’ and 3’ ends of the primer. Subsequently, the fragments were cloned into pCR Blunt II Topo using the Zero Blunt TOPO PCR Cloning kit (ThermoFisher Scientific, Waltham, MA, USA) and sequenced. After sequencing, the fragments were recloned into pGL4.10[luc2] (Promega, Madison, WI, USA). To identify the murine RGS5 minimal promoter, MBVPs or GL261 cells were transfected with the appropriate plasmids expressing the luciferase gene under the control of one of the mRGS5 promoter fragments (mRGS5-Pro1, 2, 3, 11, 12, 14, 15), and a luciferase assay was performed 48 h after transfection using the Luc-Pair™ Firefly Luciferase HS Assay Kit (GeneCopoeia, Cat# LF007). Lentivirus cloning and preparation To knockout SLUG in mural cells in vivo, we generated three different lentiviruses using the pLentiCRISPR v2 (Addgene Cat# 52961) system in which the murine SLUG specific oligonucleotide that serves as sgRNAs as well as the Cas9 enzyme, under control of the RGS5 promoter, are encoded on one plasmid [24]. Therefore, we removed the EF1α promoter in front of the Cas9 gene in pLentiCRISPR v2 and integrated the identified murine RGS5 minimal promoter fragment at this position. In a second step, we inserted three different oligos (Suppl. Table 2) that serve as sgRNAs for the knockout of murine SLUG. SLUG/SNAI2 genomic DNA and cDNA sequences were obtained from Ensembl (ENSMUSG00000022676), the CRISPR guide design tool was from Benchling, in silico off-target binding prediction was performed using Off-Spotter. The cloning strategy is shown in Suppl. Fig. 2. Lentiviral particles were generated by transfection of HEK293FT cells with the modified pLentiCRISPR v2 plasmids in combination with pLP1, pLP2 and pLP-VSVG (the latter three from Invitrogen, Darmstadt, Germany) using the Mirus TransiT-Lenti Reagent (Thermo Fisher). Viral particles collected from supernatants were concentrated and purified using PEG 6000 precipitation as described [25]. Virus titers were determined using QuickTiter™ titration kit (Cell Biolabs Inc., San Diego, USA) and were stored in aliquots at -80 °C. Animal experiments Animal work was performed in accordance with the German Animal Welfare Act and its guidelines (e.g. 3R principle) and was approved by the regional council of Tübingen (approval N05/17). RGS5-GFP pericyte reporter mice, which express GFP under the control of the RGS5 promoter, were from the Karolinska Institute (Stockholm, Sweden) and are described in detail in [26]. The mice were bred in the animal facility of the institute under SPF conditions and used at an age of 3 - 9 months. The specificity of GFP positivity to VAMCs in the brain was determined by immunofluorescence for GFP in parallel with the staining of ECs using a CD31 specific, fluorochrome coupled antibody. To determine the growth of GL261 parental (PAR) and TGF-β-knockout (TGF-β-KO) tumors, under anesthesia and analgesia the cells were stereotaxtically implanted into the right striatum of the mice at the following coordinates (1 mm rostral, 2 mm lateral of the bregma, 3 mm in depth, using the mouse brain atlas coordinates; The Mouse Brain Atlas (gaidi.ca)). The mice were intensively monitored to avoid and reduce pain. Magnetic resonance imaging (MRI) analyses using intravenously applied Gadovist (600 mg/kg of body weight) were performed thrice to determine tumor growth. To investigate alteration in tumor angiogenesis induced by the knockout of TGF-β in GBM cells, either 100.000 PAR or TGF-β-KO cells were intrastriatally implanted as described above. The mice were sacrificed when the tumors reached a median size of 1.5 - 2.5 mm as determined in a preliminary experiment. Brains were collected and processed for further analyses. To knockout SLUG in VAMCs in the tumor region, either 3x105 infectious particles of Lenti-CRISPR v2 (control virus) or a mixture of three Lenti-SLUG-KO viruses were intrastriatally injected 3 days before PAR cells were implanted using the same brain coordinates as described above. Histology and immunofluorescence Brains were collected and fixed in 4 % paraformaldehyde (PFA) for 20 hours. After subsequent dehydration via sucrose gradient, the cells were embedded in Tissue Tek (SAKURA Finetek, Alphen aan den Rijn, The Netherlands) and were stored frozen at -80 °C. Sectioning (10 µm slices) was performed on a LEICA CM3050 Cryotome using SuperFrost™ Plus slides (R. Langenbrinck GmbH). For IF staining, samples were permeabilized in PBS containing 0.2 % Triton® X-100 and blocked in 5 % bovine serum albumin (BSA)/ 0.5 % Triton® X-100. The respective primary antibody dilutions were used for overnight incubation at 4 °C: αSMA, 1:100; CD140b, 1:100; CD31, 1:100; GFP, 1:100 (all from Invitrogen, Carlsbad, CA, USA). Excess primary antibodies were removed by washing the slides thrice in PBS. The following fluorescence labeled antibodies were used for incubation at room temperature for 90 minutes: Goat anti-Chicken Alexa Fluor™ 488, 1:1000; Goat anti-Rabbit Alexa Fluor™ 405, 1:1000; Goat anti-Rabbit Alexa Fluor™ 647, 1:1000; Goat anti-Rat Alexa Fluor™ 647, 1:1000 (all from Invitrogen). After washing three times in PBS, the sections were mounted either with DAKO mounting medium or alternatively with Vectashield® antifade mounting media containing DAPI in case no AF405 secondary antibody was used for staining and were stored at 4 °C. All stained tissue samples were evaluated for the proteins of interest using the Zeiss Axio Imager with ApoTome2 (Carl Zeiss, Jena, Germany). Images were taken in different magnifications (10x, 25x, 40x and 63x) with fixed exposure settings and were processed using the ZEN blue 3.6 software (Carl Zeiss). Subsequent quantification analyses were performed using Image J (Fiji, [27]). Double and triple positive cells were counted manually. Clarity Brains were fixed and incubated in cold Hydrogen Monomers (HM) solution contain 4 % acrylamide, 4 % paraformaldehyde and 0.25 % VA-044 initiator (WAK0 Fisher Scientific, Waltham, MA, USA) for 1-2 days. Afterwards, polymerization was performed under vacuum for 2-3 h at 42 °C as previously described [28, 29]. Fixed brains were cut into 2-3 mm coronal slices. For the lipid removal, the brain slides were put in 8 % SDS/PBS and were incubated at 50 °C with continuous movement in a rotator by changing of SDS once a week until the tissue had reached a homogeneous transparency. Tissue transparency was assessed by visualization of high-contrast signals through the tissue and was reached after approximately 4 weeks. Samples were washed thrice in PBST before they were put in PBS overnight. Samples were incubated for 2 days at 37 °C in pre-incubation solution consisting of 4 % goat-serum, 1 % BSA, 0.25 % Triton and 0.01 % sodium azide. Subsequently, the brains were incubated for 5 days with the following primary antibodies diluted in pre-incubation solution at 37 °C: anti-CD31 (1:25-1:50; Abcam, Cambridge, UK) and anti-mCherry (1:500; Novus/Biotechne, Minneapolis, MN, USA). After washing the samples thrice for 2 hours in PBS containing 0.1 % Triton X, they were incubated for 2 days at 37 °C in the following secondary antibodies: goat-anti-rabbit Alexa 488, 1:200; goat-anti-chicken Alexa 546, 1:200 (both from Invitrogen) and DRAQ5 for nuclear staining (1:500, Thermo Fisher Scientific, Waltham, MA, USA). Afterwards, the samples were washed three times in PBST for 2 hours each followed by washing with either PBS containing 0.01 % sodium azide or PBST overnight. After further washing in PBS for 2 hours, samples were transferred in 80 % glycerol at 37 °C to match the refractive index within the hydrogel [30]. Confocal microscopy was performed on a Zeiss LSM 510 followed by image processing using the ZEN black software (Carl Zeiss) and Image J. Subsequent analysis was performed using Imaris (Oxford Instruments, Oxford, UK). Statistical analyses All in vitro experiments were performed at least thrice if not mentioned otherwise. For in vivo experiments, the group and sample size are indicated in the figure legends. To assume a gaussian distribution, all data received from in vitro experiments passed a normality test (Shapiro-Wilk and Tukey’s multiple comparison tests). Further statistical analyses were done with a two-tailed Student’s t-test or one-way ANOVA using GraphPad Prism 7.0 (GraphPad Inc., San Diego, CA, USA) or Excel (Microsoft Corp, Redmond, WA, USA), followed by Bonferroni correction. The results are represented as mean ± standard deviation (SD). p-values of < 0.05 are considered as statistically significant (ns: not significant; * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001). Results Characterization of the TGF-β knockout in GL261 cells in vitro and in vivo The knockout of TGF-β1 and -β2 was verified by PCR and sequencing and was found to be complete in the GL261 tumor cells, since no full length TGF-β mRNA was detected in TGF-β-knockout (TGF-β-KO) cells (Suppl. Fig. 1). As tumor angiogenesis is correlated to tumor growth and invasion, it is of importance to determine the growth rate of TGF-β-KO cells compared to that of parental (PAR) cells. In this regard, and to later on avoid effects of different tumor size of PAR and TGF-β-KO tumors on angiogenesis in our mouse GBM model, we determined proliferation as well as cell motility of the cells. As shown in Fig. 1. TGF-β-KO cells have a significantly lower capacity to proliferate and also showed lesser cell motility than PAR cells.
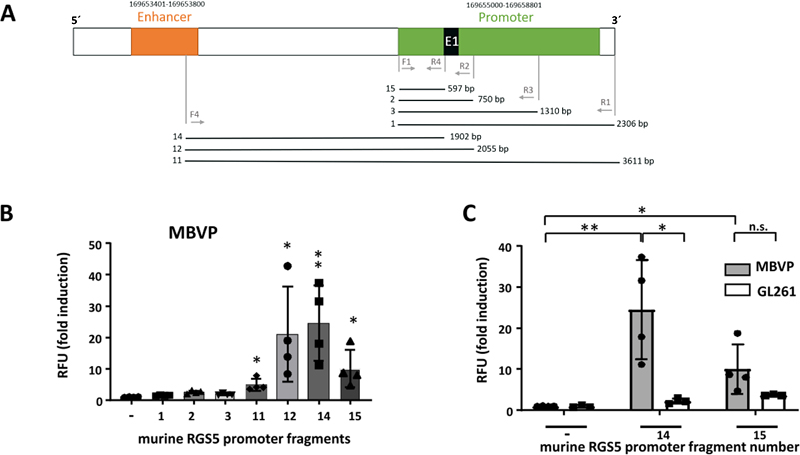
Figure 1: Characterization of GL261 TGF-β knockout cells indicates lower proliferation and cell motility upon knocking out TGF-β. Identification of the murine RGS5 minimal promoter used to knockout SLUG in tumor-associated VAMCs To knockout SLUG in vivo in our mouse GBM model, and as the murine, VAMC specific RGS5 promoter [31] has not been described in detail so far, we firstly had to identify the minimal RGS5 promoter that can be used to induce gene expression. For this, by PCR we amplified different fragments of the murine RGS promoter, cloned overlapping fragments in pGL4.10 [Luc2] and determined luciferase activity in MBVPs. Fragment 14 covers the entire region from the enhancer, ending at the start of exon 1 of the murine SLUG gene. Fragment 12 additionally includes exon 1, and fragment 15 contains 597 bp directly upstream of exon 1 (Fig. 2A). Fragments 12 and 14 showed highest luciferase activation, whilst luciferase induction by fragment 15 was only half (Fig. 2B). To determine specificity of the RGS5 promoter regions, we measured luciferase activity also in GL261 GBM cells. For fragment 14, luciferase activity in MBVPs was 25 times increased compared to the empty vector construct whilst we only saw a weak and not significant induction in GL261 cells (appr. 2-fold). For fragment 15, induction of luciferase activity was about 12x in MBVP and 4x in GL261 cells (Fig. 2C). Therefore, we chose to use fragment 14 as this fragment seemed to be the most specific part of the murine RGS5 promoter. Fragment 14 was cloned upstream of the Cas9 gene of pLentiCRISPR v2 to specifically express Cas9 in RGS5 expressing cells (pLenti-RGS5-CRISPR). The pLentiCRISPR v2 system that allows the expression of Cas9 and guide RNAs from the same vector (Suppl. Fig 2A) was used to knockout SLUG in tumor-associated mural cells in our mouse model. Three specific guide RNAs (sgRNA # 1, 4 and 5) were designed using the tool provided by Benchling (San Francisco, CA, USA). OFF-target analyses (OFF-Spotter; [32]) indicate no further targets outside of SLUG for the designed guides. The SLUG oligos were cloned into pLentiCRISPR v2 (Cas9 under control of the EF1α promoter) and target efficacy of the SLUG knockout was tested by infection of GL261 cells with these lentiviruses either alone or in combination, followed by sequencing. Using a combination of the three sgRNAs, the total editing efficiency was notably higher than for single sgRNAs and reached 85 %. InDels were found at the target sites for all three sgRNAs (Suppl. Fig. 2E). We therefore cloned sgRNAs# 1,4 and 5 into pLenti-RGS5-CRISPR, creating Lenti-SLUG-KO #1, #4 and #5, and used a combination of these three viruses to specifically knockout SLUG in GBM adjacent VAMCs in our mouse in vivo model.
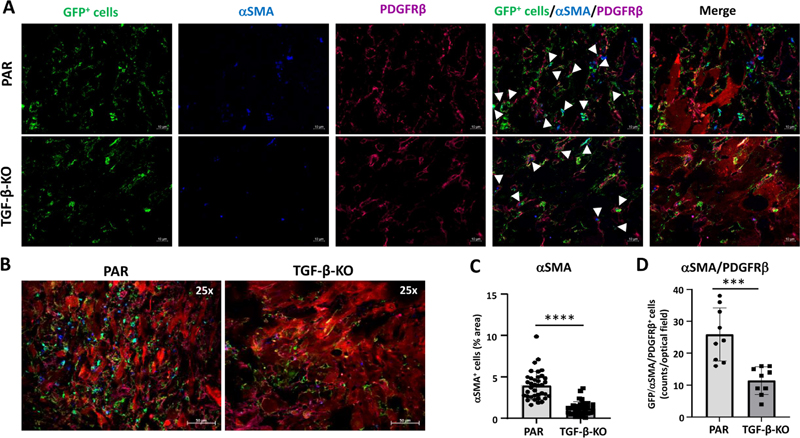
Figure 2: Identification and specificity of the murine RGS5 promoter. Knocking out TGF-β in GL261 GBMs reduces SLUG, αSMA and PDGFRβ expression in tumor-associated VAMCs and is associated with reduced tumoral VAMC and EC density In our previous studies we observed that in human brain microvascular pericytes SLUG and αSMA expression was induced by TGF-β. This observation correlates with the finding that in human GBMs, SLUG expression is associated with PDGFRβ and αSMA [16]. Therefore, in our mouse GBM model we sought to investigate whether the knockout of TGF-β in GL261 cells also influences the expression of these proteins. As tumor angiogenesis correlates with tumor size and growth, we determined the growth rate of PAR and TGF-β-KO cells and tumors in a preliminary experiment to avoid artifacts generated by different sizes of PAR and TGF-β-KO GBMs. In a preliminary experiment using MRI analyses, we observed that TGF-β-KO GL261 cells showed a delayed tumor growth also in vivo. Therefore, we determined the time point when the tumors reached a size of 1.5 to 3 mm in diameter. Material for analyses was collected when tumors were in that range of size (day 29 after implantation for PAR tumors and day 59 after implantation of TGF-β-KO-tumors (Suppl. Fig. 3A). Even if in some mice PAR tumor grew very fast (Suppl. Fig. 3C), we did not observe any significant differences in the size of PAR and TGF-β-KO tumors in the collected brain tissues in retrospective analyses (Suppl. Fig. 3 D/F). We also determined the specificity of GFP positivity in mural cells of RGS5-GFP reporter mice by CD31 and GFP staining. As indicated in Suppl. Fig. 4, GFP was only present in cells wrapping around CD31+ endothelial cells. In TGF-β-KO-GBM bearing mice, in the tumor core the number of GFP positive cells was reduced 2-fold (p < 0.01), accompanied by a highly significant reduction of SLUG (3.2 x), PDGFRβ (2.0 x) and αSMA (3.3 x) expressing cells. Notably the number of GFP+/PDGFRβ+/SLUG+ (7.4 x) and GFP+/PDGFRβ+/αSMA+ cells (2.26 x), indicating activated VAMCs, was significantly reduced (Figs. 3 and 4). Similar results, though not as pronounced, were observed in the tumor infiltration zone (Suppl. Figs. 5 and 6).
Figure 3. GFP+ VAMCs expressing SLUG and/or PDGFRβ were significantly reduced in TGF-β-KO GBMs.
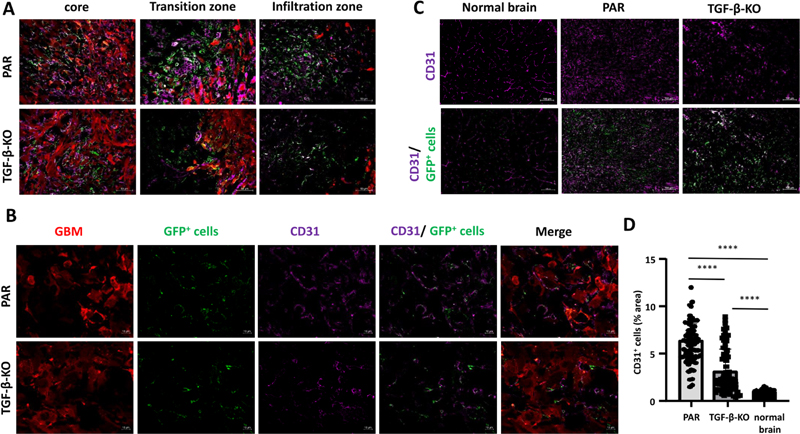
Figure 4. GFP+ VAMCs expressing αSMA and/or PDGFRβ were significantly reduced in TGF-β-KO GBMs. As GBM is a highly vascularized tumor and pericyte coverage provides vascular stability whereas TGF-β negatively influences vessel architecture, we determined vessel density in PAR and TGF-β-KO tumors. Compared to the brain parenchyma of the non-tumor bearing contralateral hemisphere, tumor areas of both, PAR and TGF-β-KO mice showed a massive enrichment in the density of CD31 cells. CD31 was used to demonstrate the presence of endothelial cells (ECs) and to determine vessel density. However, in TGF-β-KO mice, the vessel density in the tumor core was half that observed in PAR mice (Fig. 5).
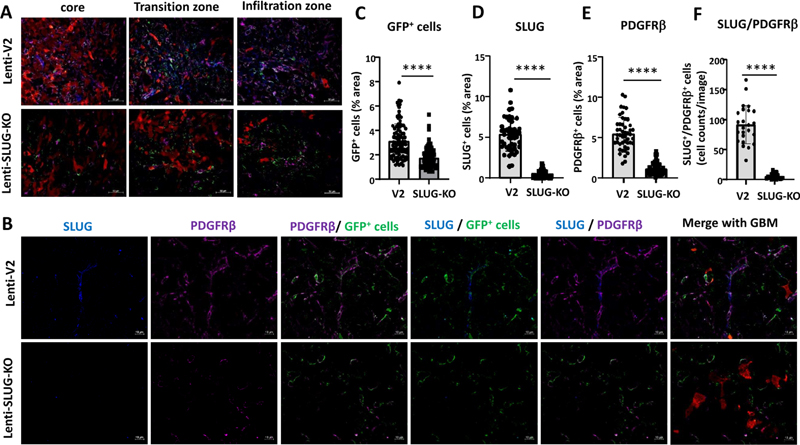
Figure 5. CD31+ ECs were significantly reduced in TGF-β-KO GBMs. Abolishing the GBM-mediated induction of SLUG expression in VAMCs reduces PDGFRβ and αSMA levels and lowers vessel density in the tumor area In a former study we identified TGF-β as an inducer of EMT transcriptional regulators such as SLUG/SNAI2 in human primary brain microvascular pericytes (HBVP). SLUG was responsible for the elevated proliferation and migration capacity of these cells, pushing HBVPs towards a more activated phenotype [16]. In this regard, we wondered whether the TGF-β-mediated effects on VAMCs and EC density observed in our mouse GBM model could be prevented by inhibiting the induction of SLUG in tumor associated VAMCs. For this, we intrastriatally injected a mixture of 3 lentiviruses that express CAS9 under the control of the RGS5 promoter as well as sgRNAs specific for murine SLUG (Lenti-SLUG-KO #1, 4 and 5; Suppl. Fig. 2) three days prior to implantation of PAR cells using the same stereotactic coordinates and collected the brains 29 days after tumor cell implantation (Suppl. Fig. 3B). To avoid artifacts that might be induced by differences in the tumor size after knocking out SLUG in mural cells in the tumor environment, we firstly analyzed the tumor size in the mice that received either the empty control lentivirus (Lenti-v2) or the mixture of Lenti-SLUG-KO viruses. However, we did not observe any significant differences in tumor size (Suppl. Fig. 3 E/G). After collecting the brains, we determined SLUG, αSMA and PDGFRβ levels in the tumor core, its transition zone, defined by the border zone between the tumor core and brain parenchyma infiltrating GBM cells, as well as in the more distant area of GBM cell infiltration. In Lenti-SLUG-KO injected mice, no SLUG expression in the tumor area was detectable anymore (Suppl. Fig 7), indicating that the SLUG-KO in VAMCs is functional. Compared to the knockout of TGF-β in GL261 cells, we observed a significant reduction, even at a higher magnitude, in the number of PDGFRβ+ (4.9-fold) and αSMA+ (10.7-fold) cells, as well as in SLUG+/PDGFRβ+/GFP+ (15.9-fold) and αSMA+/PDGFRβ+/GFP+ (11.4-fold) cells within the tumor core, the transition zone, and the infiltration zone of mice with knocked-out SLUG in tumor-associated VAMCs (Fig. 6, 7, Suppl. Fig. 8, 9).
Figure 6. GFP+ cells expressing SLUG and/or PDGFRβ were significantly reduced in mice injected with Lenti-SLUG-KO.
Figure 7. GFP+ VAMCs expressing αSMA and/or PDGFRβ were significantly reduced in mice injected with Lenti-SLUG-KO. This indicates that the activation of VAMCs (identified by the expression of PDGFRβ and/or αSMA) in the tumor core is provoked by TGF-β released from GL261 cells and the subsequent induction of SLUG expression in VAMCs located adjacent to GBM cells. Furthermore, vessel density was also significantly decreased due to the SLUG knockout in all regions of the tumor, although it did not reach the baseline levels of EC density observed in non-tumor-bearing brain regions. (Fig. 8).
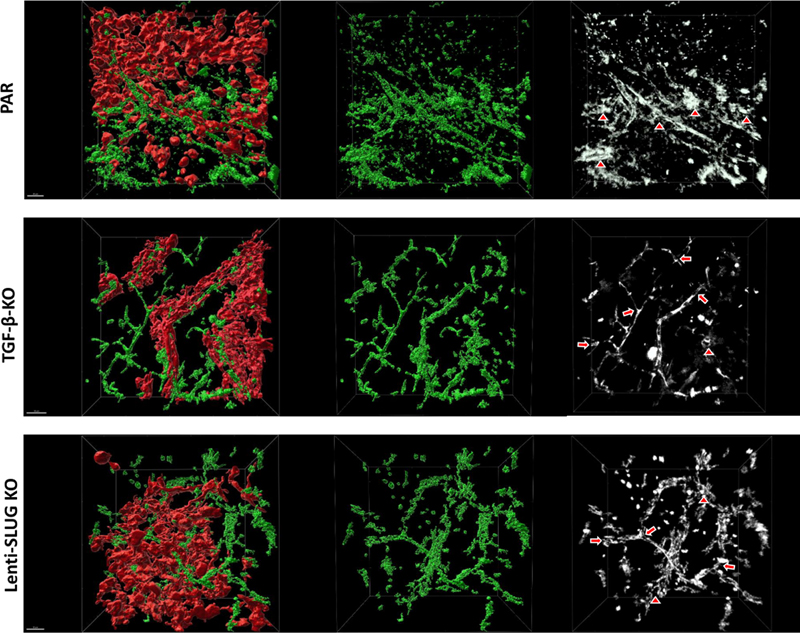
Figure 8. CD31+ ECs were significantly reduced in mice injected with Lenti-SLUG-KO. Knocking out TGF-β in GBM cells or SLUG in VAMCs attenuates pathological alterations of the intratumoral vasculature Vessels of PAR cell derived GBMs clearly displayed abundant structural alterations consisting of vascular tangles or glomeroid-like structures. They exhibited a more interconnected, "net-like" appearance and were positioned in closer proximity to each other compared to the vessels found in TGF-β-KO tumors or when SLUG induction in VAMCs was inhibited by Lenti-SLUG-KO injection. The vasculature in PAR cell derived GBMs not only showed a higher vessel density, but also greater variations. Vessels in TGF-β-KO tumors as well as in those tumors where SLUG was knocked out in VAMCs exhibited less pronounced structural vessel abnormalities and a reduced number of these cells. However, the intratumoral vessels were still chaotically organized compared to the vascular structure of the brain in the contralateral, tumor-free hemisphere of the same animal (Suppl. Fig. 10). For further detailed analysis, we conducted tissue clearing of the brain of one mouse per experimental group and performed 3D reconstruction of the tumor vasculature. The 3D vessel reconstruction strikingly shows that mouse brains bearing PAR tumor not only displayed a high vessel density but also several structural alterations such as vascular tangles or glomeroid-like structures instead of the straight vessel course seen in the contralateral brain hemisphere. We further observed that this altered and chaotic vessel structure was clearly attenuated after knocking out TGF-β in tumor cells or SLUG in VAMCs (Fig. 9; Suppl. Fig. 11).
Figure 9: Three-dimensional reconstruction of tumor-associated vessels indicates a normalized vessel structure in mice harboring TGF-β-KO GBMs as well as in mice with PAR tumors that received Lenti-SLUG-KO. Discussion GBM is a highly vascularized tumor showing altered vascular morphology with chaotically organized vessels, for which anti-angiogenic therapies were considered as promising therapeutic option. However, until now these therapies lack a remarkable increase in patient overall survival [33, 34]. Rather, vascular remodeling induced by anti-VEGF treatment results in hypoxia, is associated with excessive tumor growth with simultaneous insufficient vascularization after an initial transient vascular "normalization", but finally favoring invasiveness [35]. For a long time, it has been thought that irregular vessel development in tumors depends on the activation of ECs resulting in proliferation, migration and chaotic vascular structures. More recently, increased attention has been paid to vessel-covering pericytes and their interaction with ECs during vessel formation processes in tumors, including GBM. Brain pericytes are known to be crucial for angiogenesis, they regulate perfusion and maintain vessel structure as well as BBB integrity [36-38]. In addition, they can facilitate cerebral inflammatory processes, control the interaction between neurons and vascular cells to assist the energy demand of the brain and are an inherent part of the neurovascular unit [39]. In the healthy brain pericyte coverage is heterogeneous and dependent on the cell´s subtype. In humans, brain pericytes are classified into two subtypes with either elevated expression of transmembrane transporters (T pericytes) or of extracellular matrix (ECM) regulation genes (M pericytes). In mice, studies found two major subtypes of pericytes which either express αSMA (ensheathing pericytes) or not (mesh pericytes), the latter ones mainly located on small capillaries [40, 41]. However, within GBM, the tumor vasculature displays structural and functional abnormalities with increased and irregular pericyte coverage, that is associated with worse prognosis and accelerated tumor recurrence [42]. In our study, after knocking out TGF-β in GBM cells or SLUG in VAMCs, we saw a significant reduction of the total number of GFP+ (Fig. 3 and 6) as well as of αSMA+ cells (Fig. 4 and 7). As in our mouse model, we cannot distinguish between ensheathing and mesh pericytes, we therefore cannot clarify to the end whether lesser numbers of the one or other subtype of pericytes were observed. One further limitation of the RGS5-GFP mouse model we used in our study is that we cannot clearly distinguish between pericytes and vascular smooth muscle cells (VSMC) as also VSMCs express RGS5 and might therefore be positive for GFP [41]. In GBM, there is a pathogenic crosstalk between tumor cells and pericytes. GBM cells modify the contractile activity of pericytes, resulting in the cooption of existing blood vessels, by this supporting the expansion of the tumor [43]. In addition, glioma vessel associated pericytes support tumor growth by the induction of an immunosuppressive microenvironment [5]. Due to these tumor promoting function of pericytes, these cells have been recently identified as novel targets for the treatment of GBM [44]. In our recent studies we found that VAMCs express EMT-transcription factors like SLUG, indicating that, induced by GBM cell secreted TGF-β, these cells undergo an EMT-like "activation" process [15]. This observation was underpinned by our data that in vitro TGF-β treatment of human primary microvascular pericytes induced proliferation, cell motility and morphological changes [16] and mitigates the integrity to the BBB [17]. In our study, we were interested whether this EMT-like "activation" of glioma-associated VAMCs might also be associated with vascular alterations in vivo. In a syngeneic mouse GBM model that allows to track both GBM cells as well as RGS5 positive VAMCs, we prevented the induction of this EMT -like activation by knocking out TGF-β in the tumor cells (Suppl. Fig. 1) on the one hand, or by knocking out SLUG in tumor adjacent, RGS5 expressing VAMCs (Suppl. Fig. 7) on the other. As tumor angiogenesis is correlated to tumor size and growth, we firstly determined the growth rate of PAR and TGF-β-KO cells and tumors to avoid artifacts generated by different sizes of PAR and TGF-β-KO GBMs. After knocking out TGF-β in GL261 cells these cells showed a diminished proliferation rate and cell motility in vitro (Fig. 1), and also a delayed tumor growth in vivo. However, by using MRI we determined the time point the tumors reached a size of 1.5 to 3 mm in diameter, and therefore material for analyses was collected when tumors were in that range of size, which was also retrospectively validated in the collected material (Suppl. Fig. 3). Nevertheless, this might be a limitation of this study because even if we analyzed the vascular structure of GBMs when PAR and TGF-β-KO tumor reached the same size, we cannot completely exclude that a delayed tumor growth also influences its vascularity. For the SLUG-KO in glioma-associated VAMCs and to avoid artifacts that might be induced by knocking out SLUG in other cell types within the tumor area, we examined the expression of SLUG in the tumor area of PAR GBMs and found it to be colocalized invariably with GFP, but not with the tumor cells. In contrast, SLUG was absent in GFP+ VAMCs in those mice we injected with Lenti-SLUG-KO (Suppl. Fig. 7). However, even if CAS9 expression in Lenti-SLUG-KO was driven by the RGS5 minimal promoter, which significantly induced luciferase activity in MBVPs, but not in GL261 GBM cells (Fig. 2) we cannot completely exclude that minimal SLUG levels (below the immunofluorescence detection limit) in other cell types will be also reduced by intrastriatally applied Lenti-SLUG-KO. Previous in vitro data showed an upregulation of αSMA and PDGFRβ in HBVPs in response to TGF-β or conditioned medium from GBM cells in vitro as well as a correlation of αSMA and PDGFRβ with SLUG in human GBMs [15, 16]. In accordance with those findings, strong signals of αSMA and PDGFRβ have been detected in mice brains bearing TGF-β secreting PAR tumors (Figs. 3, 4, 6, 7). During fibrosis, αSMA and PDGFRβ are considered as pericyte activation markers, and in renal carcinoma, perivascular αSMA and PDGFRβ expression is correlated with poorer survival [45, 46]. This suggests that in vascularized high-grade glioma, glioma-associated VAMCs are pathologically activated. In parallel with the high levels of PDGFRβ and αSMA we observed in TGF-β secreting GBMs, we detected SLUG that was nearly virtually invisible in TGF-β-KO GBMs (Fig. 3, Suppl. Fig. 5). After knocking out TGF-β in the tumor cells, or SLUG in glioma-associated VAMCs, both PDGFRβ and αSMA positive cells were markedly and significantly reduced. In accordance with our previous in vitro data [16], this observation reinforces our assumption, that also in vivo in GBMs the induction of a SLUG dependent EMT-like program especially in glioma-associated VAMCs is conveyed by GBM cell secreted TGF-β. Nevertheless, apart from GBM cells, several other cell types in the CNS have been shown to secrete TGF-β, including ECs and microglial cells, which could also stimulate VAMCs to a certain amount [47]. Moreover, additional influence on this stimulation mediated by different cytokines cannot be excluded. Particularly the hypoxic microenvironment in GBM is known to be a potent inducer of EMT and morphological alterations [48]. Until now it remains unclear whether VEGF signaling also influences pericytes or other mural cells. It is known that VEGF primarily signals proliferation, survival and migration. However, a via the VEGF receptor (VEGFR)-2 leading to EC VEGF-mediated activation of VEGFR-1 has recently been shown to be not only involved in the recruitment of mural cells which is required for maturation and stabilization of neo-vessels, but also to affect vascular permeability [49, 50]. As pericytes are known to express VEGFR-1 particularly under hypoxic conditions [51], VEGF-mediated changes of pericytes towards a more mesenchymal phenotype enabling their recruitment might be considered. By preventing the EMT mediated "activation" of glioma-associated VAMCs, we observed a significant reduction of GFP+ cells in all tumor areas (Fig. 3, 6, Suppl. Fig. 5). In this regard, the source of pericytes on GBM vessels remains controversial as it has been postulated that pericytes might also originate from glioma stem like cells [52]. It has been published that GL261 cells grown as neurospheres under reduced FCS concentrations contain a population of cells with stem cell characteristics [53]. To avoid stemness, in our study GL216 cells were grown in 10% FCS before implantation. However, we cannot exclude that in vivo in the growing tumor, stem cell characteristics were induced in the tumor cells. As GL261 cells do not express GFP and therefore GL261 derived pericytes cannot be detected by our approach, we cannot exclude that a small population of GL261 with stem cell characteristics might develop into pericytes. However, we did not observe any cells that are double positive of mCherry and a mural cell marker such as αSMA or, in TGF-β-secreting PAR tumors, positive for SLUG (Fig. 3 and 4). Besides, it has been published that in GL261 tumor bearing mice the vast majority of pericytes within GBMs are definitely endogenous, host-derived cells originating from either mesenchymal or neural crest cells [54]. Hence, we believe that our mouse GBM model is suitable to investigate the TGF-β/SLUG mediated modulation of glioma-associated VAMCs in vivo. Vascularization not only involves EC proliferation, migration, branching and anastomosis, it also requires adequate pericyte coverage of vascular sprouts for vessel stabilization and maturation (for review see [55]). At least in GL261 tumors, the knockout of TGF-β or the prevention of a SLUG-mediated EMT-like activation of VAMCs in the tumor environment, reduces the number of GFP+ cell numbers as well as that of CD31+ cells, suggesting that not only ECs, but also VAMCs are main cells that constitute the multiplicity and altered morphology of tumor microvessels. Physiologically, pericytes constitute a monolayer surrounding ECs (for review see [56]). However, even if the vessel coverage with GFP+ VAMCs in the tumor area was heterogeneous and areas with multilayers of overlapping GFP+ cells can be distinguished from vessels that display almost no coverage, the irregular covering correlated significantly with elevated SLUG, PDGFRβ and αSMA levels. This underpins that the shift towards a more mesenchymal phenotype of glioma-associated VAMCs and their subsequent activation might contribute to the development of vascular alterations in GBM. Unlike the tight association of GFP+ VAMCs and ECs that can be observed in normal vessels, cells with elevated PDGFRβ and αSMA levels display an abnormal separation, but also multi-layered coverage of vessels (Suppl. Fig. 10). As the direct contact and communication between pericytes and ECs are critical for maintenance of cerebrovascular stability and blood-brain-barrier function, the observed loose association of GFP+ cells to tumor vessels might result in vascular dysfunction. Aberrations in pericyte-EC signaling are already known to contribute to tumor angiogenesis [57, 58]. Apart from abnormal pericyte coverage, tumor vessels displayed various structural abnormalities. The observed high amount of "EMT-activated-state" GFP+ glioma-associated VAMCs in PAR tumors was paralleled by an increased vascular density and vascular abnormalities which suggests an important role of activated pericytes in glioma-associated neoangiogenic processes. Tumor blood vessels displayed inconsistent diameter and uneven shape with abnormal bulges, blind ends and glomeroid-like structures (Fig. 5, 8, 9, Suppl. Fig. 10, 11). In GBMs, structural and functional abnormalities of peritumoral vessels are often associated with an inconsistent, disrupted BBB that may constitute a main hurdle for targeting GBM by limiting adequate drug delivery. Pericytes, being important contributors to the establishment and maintenance of BBB integrity by regulating tight and adherens junctions as well as transcytosis across endothelial cells [59], can be functionally modified by GBM cells [60]. Via the TGF-β-mediated induction of EMT-factors in VAMCs, GBM cells are able to induce proliferation and cell motility [15, 16] and notably change the metabolic behavior of pericytes. Schumacher et al. demonstrated that TGF-β treatment of HBVPs results in BBB disruption at least in vitro. Additional metabolomic and transcriptomic analyses underscored that the TGF-β-mediated functional and metabolic changes in pericytes are closely connected with their role during angiogenic processes [17]. In combination with the lower mural cell activation state and the less chaotic vessel structure, we observed after preventing the induction of SLUG expression in glioma associated VAMCs in vivo, our findings suggest that the BBB integrity might also be affected. However, this has to be further evaluated for example by measuring the uptake of contrast agents using small animal MRI. Conclusions Induced by GBM cell secreted TGF-β, microvascular brain pericytes undergo an EMT-like "activation" process, indicated by an induction of the EMT-associated expressional regulator SLUG. In vitro, this phenomenon is associated with elevated proliferation, cell motility and morphological changes. In our study, we demonstrate that an EMT-like "activation" of glioma-associated, RGS5 expressing VAMCs is also associated with vascular malformation. Both the knockout of TGF-β in glioma cells and the prevention of SLUG-induction in glioma-associated VAMCs mitigated the amount of activated pericytes or VAMCs in the tumor core, as shown by a reduced expression of activation markers such as PDGFRβ or αSMA. In addition, the prevention of this activation diminished the total amount of GFP+ VAMCs covering the tumor vasculature and also, at least partially, led to a restoration of the chaotic vessel structure towards a more normal vascular morphology. In summary, our findings suggest that the induction of an EMT-like program in glioma-associated VAMCs is the prominent cause of vessel abnormalities frequently observed in GBM. Acknowledgment We would like to acknowledge Dr. Olga Oleksiuk of the HIH-CIN Imaging Cluster for her support in all microscopy and microanalysis experiments. We also like to thank Ulrich Mattheus for his support in the clarity experiments. Michel Mittelbronn would like to thank the Luxembourg National Research Fond (FNR) for the support (FNR PEARL P16/BM/11192868 grant). Authors' contributions UN has designed and supervised the study, has full access to all data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. UN and LM wrote the manuscript. MM performed critical review of the manuscript. LM, KR, HE, ME, AEA, MM, JJG and MK performed experiments and analyzed data. Institutional Review Board Statement All research meets ethical guidelines and adheres to the legal requirements of the study country. Animal work was performed in accordance with the German Animal Welfare Act and its guidelines and was approved by the Regional Council of Tübingen (approval N7/17). Data Availability Statement The datasets and material generated and/or analyzed during the current study are available on demand. Conflict of Interest The authors declare no conflicts of interest. All coauthors have reviewed and approved the contents of the manuscript and that the requirements for authorship have been met. Institutional Review Board Statement All research meets ethical guidelines and adheres to the legal requirements of the study country. Animal work was performed in accordance with the German Animal Welfare Act and its guidelines and was approved by the Regional Council of Tübingen (approval N5/17). Funding Statement This study has been funded by the IZKF Promotionsprogramm of the Medical Faculty, University of Tübingen. References 1. Schaff, L.R. and I.K. Mellinghoff, Glioblastoma and Other Primary Brain Malignancies in Adults: A Review. JAMA, 2023. 329(7): p. 574-587. https://doi.org/10.1001/jama.2023.0023 2. Stupp, R., et al., Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA : the journal of the American Medical Association, 2017. 318(23): p. 2306-2316. https://doi.org/10.1001/jama.2017.18718 3. Hochberg, F.H., et al., Glioma diagnostics and biomarkers: an ongoing challenge in the field of medicine and science. Expert Rev Mol Diagn, 2014. 14(4): p. 439-52. https://doi.org/10.1586/14737159.905202 4. Ochs, K., et al., Immature mesenchymal stem cell-like pericytes as mediators of immunosuppression in human malignant glioma. Journal of neuroimmunology, 2013. 265(1-2): p. 106-16. https://doi.org/10.1016/j.neuroim.2013.09.011 5. Sena, I.F.G., et al., Glioblastoma-activated pericytes support tumor growth via immunosuppression. Cancer Med, 2018. 7(4): p. 1232-1239. https://doi.org/10.1002/cam4.1375 6. Waziri, A., Glioblastoma-derived mechanisms of systemic immunosuppression. Neurosurg Clin N Am, 2010. 21(1): p. 31-42. https://doi.org/10.1016/j.nec.2009.08.005 7. Himes, B.T., et al., Immunosuppression in Glioblastoma: Current Understanding and Therapeutic Implications. Front Oncol, 2021. 11: p. 770561. https://doi.org/10.3389/fonc.2021.770561 8. Gong, L., et al., TGF-beta links glycolysis and immunosuppression in glioblastoma. Histol Histopathol, 2021. 36(11): p. 1111-1124. https://doi.org/10.14670/HH-18-366 9. Auffinger, B., et al., The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert review of neurotherapeutics, 2015. 15(7): p. 741-52. https://doi.org/10.1586/14737175.2015.1051968 10. D'Alessio, A., et al., Pathological and Molecular Features of Glioblastoma and Its Peritumoral Tissue. Cancers (Basel), 2019. 11(4). https://doi.org/10.3390/cancers11040469 11. Das, S. and P.A. Marsden, Angiogenesis in glioblastoma. N Engl J Med, 2013. 369(16): p. 1561-3. https://doi.org/10.1056/NEJMcibr1309402 12. Birner, P., et al., Vascular patterns in glioblastoma influence clinical outcome and associate with variable expression of angiogenic proteins: evidence for distinct angiogenic subtypes. Brain Pathol, 2003. 13(2): p. 133-43. https://doi.org/10.1111/j.1750-3639.2003.tb00013.x 13. Jackson, S., et al., Blood-brain barrier pericyte importance in malignant gliomas: what we can learn from stroke and Alzheimer's disease. Neuro Oncol, 2017. 19(9): p. 1173-1182. https://doi.org/10.1093/neuonc/nox058 14. Sattiraju, A. and A. Mintz, Pericytes in Glioblastomas: Multifaceted Role Within Tumor Microenvironments and Potential for Therapeutic Interventions. Adv Exp Med Biol, 2019. 1147: p. 65-91. https://doi.org/10.1007/978-3-030-16908-4_2 15. Mader, L., et al., Pericytes/vessel-associated mural cells (VAMCs) are the major source of key epithelial-mesenchymal transition (EMT) factors SLUG and TWIST in human glioma. Oncotarget, 2018. 9(35): p. 24041-24053. https://doi.org/10.18632/oncotarget.25275 16. Wirsik, N.M., et al., TGF-beta activates pericytes via induction of the epithelial to mesenchymal transition protein SLUG in glioblastoma. Neuropathol Appl Neurobiol, 2021. 47(6):p. 768-780. https://doi.org/10.1111/nan.12714 17. Schumacher, L., et al., TGF-Beta Modulates the Integrity of the Blood Brain Barrier In Vitro, and Is Associated with Metabolic Alterations in Pericytes. Biomedicines, 2023. Special Issue "Angiogenesis in Heath and Disease 2.0", (11, 214): p. 1-19. https://doi.org/10.3390/biomedicines11010214 18. Ueda, R., et al., Systemic inhibition of transforming growth factor-beta in glioma-bearing mice improves the therapeutic efficacy of glioma-associated antigen peptide vaccines. Clin Cancer Res, 2009. 15(21): p. 6551-9. https://doi.org/10.1158/1078-0432.CCR-09-1067 19. Ausman, J.I., W.R. Shapiro, and D.P. Rall, Studies on the chemotherapy of experimental brain tumors: development of an experimental model. Cancer Res, 1970. 30(9): p. 2394-400. 20. Oh, S.A., A. Seki, and S. Rutz, Ribonucleoprotein Transfection for CRISPR/Cas9-Mediated Gene Knockout in Primary T Cells. Curr Protoc Immunol, 2019. 124(1): p. e69. https://doi.org/10.1002/cpim.69 21. Stemmer, M., et al., CCTop: An Intuitive, Flexible and Reliable CRISPR/Cas9 Target Prediction Tool. PLoS One, 2015. 10(4): p. e0124633. https://doi.org/10.1371/journal.pone.0124633 22. Bartussek, C., U. Naumann, and M. Weller, Accumulation of mutant p53(V143A) modulates the growth, clonogenicity, and radiochemosensitivity of malignant glioma cells independent of endogenous p53 status. Exp Cell Res, 1999. 253(2): p. 432-9. https://doi.org/10.1006/excr.1999.4654 23. Schotterl, S., et al., Viscumins functionally modulate cell motility-associated gene expression. Int J Oncol, 2017. 50(2): p. 684-696. https://doi.org/10.3892/ijo.2017.3838 24. Shalem, O., et al., Genome-scale CRISPR-Cas9 knockout screening in human cells. Science, 2014. 343(6166): p. 84-87. https://doi.org/10.1126/science.1247005 25. Fricano-Kugler, C.J., et al., Designing, Packaging, and Delivery of High Titer CRISPR Retro and Lentiviruses via Stereotaxic Injection. J Vis Exp, 2016(111). https://doi.org/10.3791/53783 26. Nisancioglu, M.H., et al., Generation and characterization of rgs5 mutant mice. Molecular and cellular biology, 2008. 28(7): p. 2324-31. https://doi.org/10.1128/MCB.01252-07 27. Schindelin, J., et al., Fiji: an open-source platform for biological-image analysis. Nature methods, 2012. 9(7): p. 676-82. https://doi.org/10.1038/nmeth.2019 28. Neckel, P.H., et al., Large-scale tissue clearing (PACT): Technical evaluation and new perspectives in immunofluorescence, histology, and ultrastructure. Sci Rep, 2016. 6: p. 34331. https://doi.org/10.1038/srep34331 29. Chung, K. and K. Deisseroth, CLARITY for mapping the nervous system. Nat Methods, 2013. 10(6): p. 508-13. https://doi.org/10.1038/nmeth.2481 30. Du, H., et al., Advances in CLARITY-based tissue clearing and imaging. Exp Ther Med, 2018. 16(3): p. 1567-1576. https://doi.org/10.3892/etm.2018.6374 31. Bondjers, C., et al., Transcription profiling of platelet-derived growth factor-B-deficient mouse embryos identifies RGS5 as a novel marker for pericytes and vascular smooth muscle cells. The American journal of pathology, 2003. 162(3): p. 721-9. https://doi.org/10.1016/S0002-9440(10)63868-0 32. Pliatsika, V. and I. Rigoutsos, "Off-Spotter": very fast and exhaustive enumeration of genomic lookalikes for designing CRISPR/Cas guide RNAs. Biol Direct, 2015. 10: p. 4. https://doi.org/10.1186/s13062-015-0035-z 33. Hamza, M.A., et al., Survival outcome of early versus delayed bevacizumab treatment in patients with recurrent glioblastoma. J Neurooncol, 2014. 119(1): p. 135-40. https://doi.org/10.1007/s11060-014-1460-z 34. Gilbert, M.R., et al., A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med, 2014. 370(8): p. 699-708. https://doi.org/10.1056/NEJMoa1308573 35. Keunen, O., et al., Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc Natl Acad Sci U S A, 2011. 108(9): p. 3749-54. https://doi.org/10.1073/pnas.1014480108 36. Hill, J., et al., Emerging roles of pericytes in the regulation of the neurovascular unit in health and disease. J Neuroimmune Pharmacol, 2014. 9(5): p. 591-605. https://doi.org/10.1007/s11481-014-9557-x 37. Sweeney, M.D., S. Ayyadurai, and B.V. Zlokovic, Pericytes of the neurovascular unit: key functions and signaling pathways. Nat Neurosci, 2016. 19(6): p. 771-83. https://doi.org/10.1038/nn.4288 38. Liu, S., et al., The role of pericytes in blood-brain barrier function and stroke. Current pharmaceutical design, 2012. 18(25): p. 3653-62. https://doi.org/10.2174/138161212802002706 39. Brown, L.S., et al., Pericytes and Neurovascular Function in the Healthy and Diseased Brain. Front Cell Neurosci, 2019. 13: p. 282. https://doi.org/10.3389/fncel.2019.00282 40. Yang, A.C., et al., A human brain vascular atlas reveals diverse mediators of Alzheimer's risk. Nature, 2022. 603(7903): p. 885-892. https://doi.org/10.1038/s41586-021-04369-3 41. Bohannon, D.G., D. Long, and W.K. Kim, Understanding the Heterogeneity of Human Pericyte Subsets in Blood-Brain Barrier Homeostasis and Neurological Diseases. Cells, 2021. 10(4). https://doi.org/10.3390/cells10040890 42. Zhang, X.N., et al., Pericytes augment glioblastoma cell resistance to temozolomide through CCL5-CCR5 paracrine signaling. Cell Res, 2021. 31(10): p. 1072-1087. https://doi.org/10.1038/s41422-021-00528-3 43. Caspani, E.M., et al., Glioblastoma: a pathogenic crosstalk between tumor cells and pericytes. PloS one, 2014. 9(7): p. e101402. https://doi.org/10.1371/journal.pone.0101402 44. Guerra, D.A.P., et al., Targeting glioblastoma-derived pericytes improves chemotherapeutic outcome. Angiogenesis, 2018. 21(4): p. 667-675. https://doi.org/10.1007/s10456-018-9621-x 45. Chen, Y.T., et al., Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int, 2011. 80(11): p. 1170-81. https://doi.org/10.1038/ki.2011.208 46. Frodin, M., et al., Perivascular PDGFR-beta is an independent marker for prognosis in renal cell carcinoma. Br J Cancer, 2017. 116(2): p. 195-201. https://doi.org/10.1038/bjc.2016.407 47. Rustenhoven, J., et al., TGF-beta1 regulates human brain pericyte inflammatory processes involved in neurovasculature function. J Neuroinflammation, 2016. 13: p. 37. https://doi.org/10.1186/s12974-016-0503-0 48. Iwadate, Y., Epithelial-mesenchymal transition in glioblastoma progression. Oncology letters, 2016. 11(3): p. 1615-1620. https://doi.org/10.3892/ol.2016.4113 49. Uemura, A., et al., VEGFR1 signaling in retinal angiogenesis and microinflammation. Prog Retin Eye Res, 2021. 84: p. 100954. https://doi.org/10.1016/j.preteyeres.2021.100954 50. Cicatiello, V., et al., Powerful anti-tumor and anti-angiogenic activity of a new anti-vascular endothelial growth factor receptor 1 peptide in colorectal cancer models. Oncotarget, 2015. 6(12): p. 10563-76. https://doi.org/10.18632/oncotarget.3384 51. Eilken, H.M., et al., Pericytes regulate VEGF-induced endothelial sprouting through VEGFR1. Nat Commun, 2017. 8(1): p. 1574. https://doi.org/10.1038/s41467-017-01738-3 52. Cheng, L., et al., Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell, 2013. 153(1): p. 139-52. https://doi.org/10.1016/j.cell.2013.02.021 53. Yi, L., et al., Implantation of GL261 neurospheres into C57/BL6 mice: a more reliable syngeneic graft model for research on glioma-initiating cells. Int J Oncol, 2013. 43(2): p. 477-84. https://doi.org/10.3892/ijo.2013.1962 54. Svensson, A., et al., Endogenous brain pericytes are widely activated and contribute to mouse glioma microvasculature. PloS one, 2015. 10(4): p. e0123553. https://doi.org/10.1371/journal.pone.0123553 55. Girolamo, F., et al., Central Nervous System Pericytes Contribute to Health and Disease. Cells, 2022. 11(10). https://doi.org/10.3390/cells11101707 56. Berthiaume, A.A., et al., Pericyte Structural Remodeling in Cerebrovascular Health and Homeostasis. Front Aging Neurosci, 2018. 10: p. 210. https://doi.org/10.3389/fnagi.2018.00210 57. Schiffer, D., et al., Glioblastoma niches: from the concept to the phenotypical reality. Neurol Sci, 2018. 39(7): p. 1161-1168. https://doi.org/10.1007/s10072-018-3408-0 58. Schiffer, D., et al., Glioblastoma: Microenvironment and Niche Concept. Cancers, 2018. 11(1). https://doi.org/10.3390/cancers11010005 59. Liebner, S., et al., Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol, 2018. 135(3): p. 311-336. https://doi.org/10.1007/s00401-018-1815-1 60. Thanabalasundaram, G., et al., The impact of pericytes on the blood-brain barrier integrity depends critically on the pericyte differentiation stage. Int J Biochem Cell Biol, 2011. 43(9): p. 1284-93. https://doi.org/10.1016/j.biocel.2011.05.002
Copyright: © 2024 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |