|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Free Neuropathology 4:17 (2023) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Original Paper |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Pathological perspectives in pilocytic astrocytomas: Extent of resection as the sole critical factor for recurrence-free survival, and the challenge of evaluating conclusions derived from limited data |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Ibrahim Kulac1, Irem Yenidogan2, Banu Oflaz Sozmen2,3, Arzu Baygul4, Soonmee Cha5, Melike Pekmezci6, Tarik Tihan6 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Corresponding author: |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Submitted: 17 September 2023 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Keywords: Astrocytoma, Entity, Circumscribed gliomas, Glioma, Juvenile pilocytic astrocytoma, Pilocytic astrocytoma, Piloid, Pilomyxoid, Tumor type |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Abstract
Introduction: Pilocytic astrocytoma (PA) is one of the most common primary intracranial neoplasms in childhood with an overall favorable prognosis. Despite decades of experience, there are still diagnostic and treatment challenges and unresolved issues regarding risk factors associated with recurrence, most often due to conclusions of publications with limited data. We analyzed 499 patients with PA diagnosed in a single institution over 30 years in order to provide answers to some of the unresolved issues. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
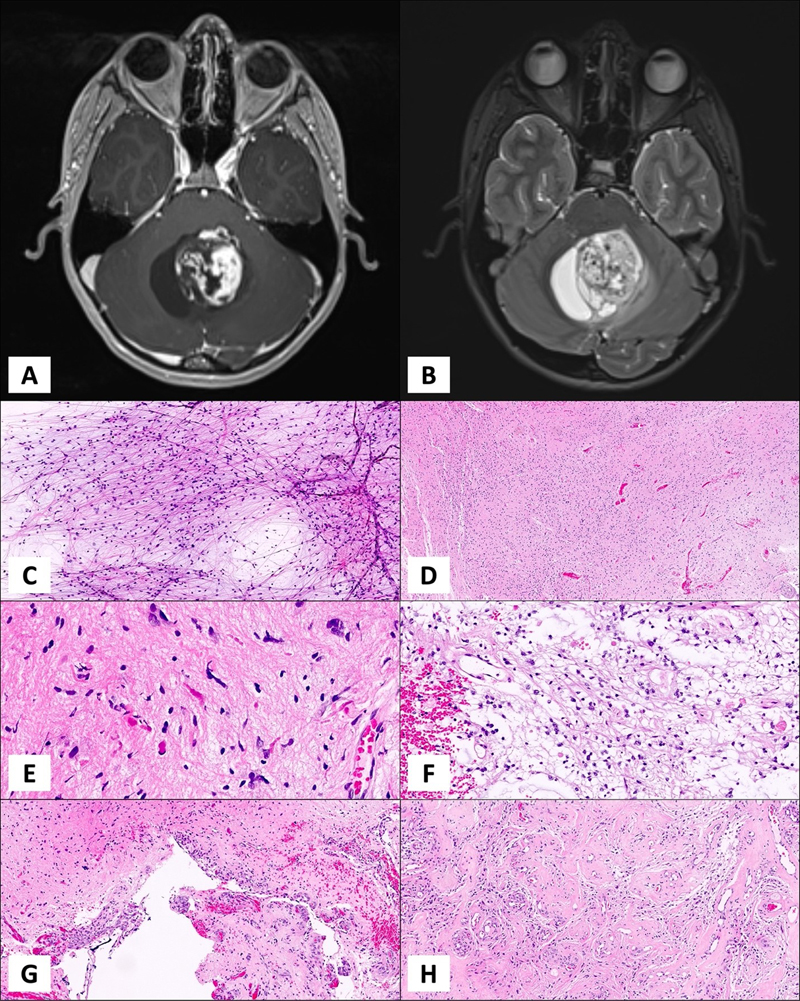
Introduction Pilocytic astrocytoma (PA) is a circumscribed astrocytoma with classic histologic features such as biphasic compact and loose growth patterns, piloid cytology, and low proliferative activity, with or without Rosenthal fibers and/or eosinophilic granular bodies [1]. The essential criteria adopted by the World Health Organization (WHO) 2021 also defines PA as a “piloid astrocytic neoplasm with a solitary MAPK pathway alteration, such as KIAA1549::BRAF fusion” [2]. Over the last century, beginning with the first use of the word “pilocytic” [3] WHO classification schemes defined PA as a clinically, radiologically, pathologically, and most recently, a molecularly distinct entity. Typical radiological and histological features are presented in Figure 1.
Figure 1. A cystic tumor with mural nodule in posterior fossa on axial T1 post-contrast (A) and T2 (B) weighted MRI images. An intraoperative smear slide of a PA case with plenty of piloid cells distributed on a glial background (C). Hematoxylin & eosin image of a demonstrative PA case with an overall fibrillary appearance (D), Closer view with plenty of Rosenthal fibers, classical finding in PAs (E). Myxoid, loose areas are another common finding in PA (F). Linear glomeruloid vascular structures are commonly observed (G) along with hyalinized, thick vessels (H). PA can be observed at any age with a reported incidence rate of approximately 0.84 per 100,000 [4-6], and has a favorable outcome [5]. Despite being the most common glioma among the pediatric population, overall rarity of this neoplasm makes it an orphan disease as well as a “chronic disease” affecting patients and families for many years, creating challenges in its management [7-11]. Coding of PAs in tumor registries has been problematic in recent years due to their designation as “malignant” in some countries. This change was made in order to capture these tumors in cancer registries [12]. Consequently, incidence of benign or malignant pediatric brain tumors show significant variations over time in epidemiological analyses [13]. We are not sure whether designating PAs as “malignant” in order to capture them in registries is a good idea, or whether this practice should be abandoned to bring more clarity and uniformity to the issue worldwide, but risk losing their identification in cancer statistics. PA is most commonly located in the posterior fossa, specifically in cerebellar hemispheres. Other common locations include hypothalamic/chiasmatic region and the optic nerve, the latter being particularly common in the setting of Neurofibromatosis 1 syndrome [14]. Thalamic, cerebral, and spinal tumors are distinctly less common [5]. Some authors suggested that there were prognostic differences among PAs in different locations that could not be explained simply by differences in clinical variables [15-17]. It is not clear whether different locations are associated with different outcomes beyond access to gross total resection, and whether tumor location is associated with different driver mutations and treatment response. In addition to location, some studies suggest that age is important determinant of outcome, and adult and pediatric PAs do not have similar prognoses [18-21]. PAs can have diverse morphological features, some of which may result in misclassification of some tumors as diffuse gliomas. Some PAs with the so-called “diffuse pilocytic pattern” may be easily misdiagnosed as diffuse gliomas [22]. Conversely, other entities may also be misclassified as PA and some high-grade tumors with remote resemblance to PA may be erroneously classified as tumors with piloid features or simply anaplastic PA [23]. High-grade histological features in otherwise typical PAs are also problematic and the current WHO classification scheme did not find sufficient evidence to define an anaplastic subtype [24]. However, there is clear evidence that rare PAs behave aggressively and some of these tumors may be justifiably designated as high-grade gliomas [25]. On the other hand, some worrisome histological features in PA have not been associated with adverse prognosis [26-28]. Additional studies and observations are needed to validate a high-grade subtype of PA and its definition. Currently, the only recognized subtype of PA is the pilomyxoid astrocytoma (PMA), that appears to be more aggressive compared to typical PAs [29]. However, in the recent editions of the WHO classification of central nervous system (CNS) tumors, the working groups decided that the data for recognizing this subtype as more aggressive were not sufficient to designate a specific WHO grade for PMA [15, 29, 30]. Further studies are needed for the pilomyxoid tumors to allow their grading and prognostication. Recent genomic studies outline the molecular landscape of PAs as tumors with a very stable genome, carrying less than 0.1 mutations per megabase [31, 32]. Mitogen activated protein kinase (MAPK) pathway is the most commonly affected pathway in PAs. The most common genetic alteration is the BRAF internal tandem duplication resulting with a fusion of a neighboring gene, KIAA1549. Other alterations of the MAPK pathway, such as other BRAF fusions, mutations in BRAF, RAS or NF1 were also reported [2, 33-39]. While the type of MAPK alterations has not been associated with different outcomes, it is not clear whether the cumulative effect of multiple genetic alterations may lead to a more aggressive behavior in PAs. Some studies implicated molecular alterations such as loss of CDKN2A, gain of chromosome 7 and loss of chromosome 17q as being associated with worse outcome, but none of these studies were validated in large series, prospective trials or by more than one group [25, 40]. Several clinical and biological markers reportedly influence prognosis in patients with PA, but these parameters, with the exception of extent of resection, have not been consistently found as independent variables [26-28, 41]. We also suggested that access to healthcare is a key factor in the outcome of patients with PA, most likely unrelated to tumor biology [42]. In this retrospective study, we report our experience with 499 PAs diagnosed and treated in a single institution over the last three decades in order to bring clarity to some of the uncertainties mentioned above, and to further demonstrate the need for collaborative studies and larger series to better understand factors associated with outcome. Material and Methods Patient selection All patients diagnosed as PA or PMA between 1989 and 2019, were retrieved from the pathology archives of the Department of Pathology. The inclusion criteria were as follows: 1- all patients diagnosed and/or treated at our institution between 1989 and 2019, and 2- sufficient clinical information to include into the database, and 3- sufficient pathology material available for review and diagnosis, and 4- the diagnosis of PA or PMA upon review of the available material. The database search included a series of keywords for final diagnoses, and the search was conducted at two separate occasions, which yielded nearly identical results with a rare exception due to delay in the database registry. Clinical and radiological information were obtained from the hospital electronic information systems or from the patient charts for older cases. All relevant clinical information necessary for the purposes of our analyses were collected in an anonymized fashion and a research database was created. Radiological reports were also reviewed to ensure that radiological impression was consistent with PA. Extent of resection was obtained from the operative reports and was reported as either gross total resection (GTR) or subtotal resection (STR). Any level of resection less than GTR was considered STR. Recurrence was defined as worsening of clinical symptoms attributed to tumor growth. Recurrence-free survival (RFS) was calculated between the date of the initial surgery and the first record of clinical worsening in the chart. Overall survival was calculated as the time between the initial surgery and death due to tumor, and all others were censored. The cut-off for the follow-up time was January 2022. Patients who could not be identified in the system by that date were considered lost to follow-up. This study was approved by the University of California San Francisco Institutional Review Board (IRB approval no. CHR 10-01252). Pathological evaluation All available pathology material from all cases were reviewed by two of the authors (IK, TT) and one of the authors (MP) contributed to the pathological review process for the recent cases during the last decade. PA and PMA diagnoses were based on the essential and desirable criteria proposed by the WHO 2021 classification scheme. Pertinent histopathological features were recorded. Immunohistochemical studies were performed as a part of the routine work-up of cases to establish diagnosis and further characterize the histological features. BRAF V600 mutation analysis by real time PCR A total of 222 patient samples were submitted to BRAF V600E mutational analysis by real time PCR as a part of their routine diagnosis. Real time PCR was performed according to published methodology at the Clinical Cancer Genomics Laboratory that operates under a CLIA license. Appropriate controls and quality assurance parameters have been established at this laboratory (https://genomics.ucsf.edu/content/braf-mutation-testing-including-v600e). BRAF duplication / KIAA1549::BRAF fusion analysis In limited number of cases, BRAF duplication only was investigated via fluorescence in-situ hybridization (FISH) as previously described as a part of the patients’ routine diagnostic work-up at the Clinical Cancer Genomics Laboratory (https://genomics.ucsf.edu/content/braf-rearrangement-fish). Analysis of molecular alterations and copy number variations (CNV) Capture-based next-generation DNA sequencing (NGS) was performed at the UCSF Clinical Cancer Genomics Laboratory (also referred as UCSF500 NGS assay) in 106 patients as a part of their routine diagnostic work-up according to protocols described previously [43]. Further information on the specifics of the UCSF500 NGS platform is available at the CCGL website (https://genomics.ucsf.edu/content/ucsf-500-cancer-gene-panel-test-ucsf500-uc500). Statistical analysis Statistical analysis was performed using the IBM SPSS Statistics for Windows (IBM Corp., Version 28.0, released 2021, Armonk, NY: IBM Corp). The normality of continuous variables was investigated by Shapiro-Wilk’s test. Descriptive statistics were presented using mean, standard deviation, median and interquartile range. Categorical variables were expressed by using frequencies (n) and percentages (%). To compare categorical variables Chi-Square test (or Fisher exact test/Yates continuity correction as appropriate) was used. The recurrence-free survival was evaluated by Kaplan-Meier method and the median survival times were compared by log rank test. The associations between clinicopathologic features and the recurrence free survival were evaluated by Cox regression model. The cut off for statistical significance was set as p<0.001. Results Demographic and clinical features Among a total of 528 patients identified from the database with the diagnosis of PA or PMA between 1989-2019, 499 patients fulfilled the inclusion criteria and were included in this study. Mean age at the diagnosis was 15.5 years (range: 3.4 months-72 years) and the median age was 12 years. There were 276 females (55.3%) and 223 males (44.7%). The patients were grouped into three age categories; 286 patients were <14 years old, 77 patients were between 14-21 years old, and 136 cases were >21 years old. The median follow-up was 78.5 months (1 – 580 months) and 47 (9.4%) patients had a follow-up period less than 12 months. In addition, 178 patients had >5 years and 114 patients had >10 years of follow-up. Non-surgical treatment data were available on approximately half (n=298) of the cases and among them, 82 patients have received chemotherapy and 63 have received radiotherapy. Six patients died during the follow-up period, but detailed analysis of their disease course could not directly establish cause of death as being due to tumor. All six patients initially underwent subtotal resections, received radiotherapy and multiple chemotherapy regimens, and had significant comorbidities (see Table 1). In most cases, there was no clear tumor growth, and the disease course was complicated by additional factors. Table 1. Clinical details of deceased patients
• Radiation-associated necrosis was confirmed on radioimaging as well as pathological studies of the recurrent tumors Tumor recurrence information was available for 321 patients, of which 109 suffered at least one recurrence (34%, 109 of 321 cases). Major demographic and clinical features are presented in table 2. Tumor localization Among the cases where the localization information was available (n=494), 259 (52.6%) were in the posterior fossa, 208 (42.3%) were supratentorial (includes hemispheric, hypothalamic/chiasmatic, intraventricular and optic nerve tumors), and 25 (5.1%) were in the spinal cord. Two patients had multifocal tumors at diagnosis, and the exact information on location was not available for 5 patients. Table 2. Major demographic and clinical features of patients
n: Total number of patients; STR: subtotal resection; GTR: gross total resection. Among 208 supratentorial tumors, 108 (51.9%) were in the cerebral hemispheres, 79 (38%) were in the hypothalamic/chiasmatic region, 6 (2.9%) were intraventricular, and 15 (7.2%) were involving the optic nerve. While there was a suggestion that posterior fossa tumors were less likely to recur compared with hypothalamic/chiasmatic tumors, there was no statistical significance (p=0.043) partly due to the small number of cases for each location category when controlled for other variables. Even after regrouping cases into “hypothalamo-suprachiasmatic+brainstem" and "other locations", we did not find statistically significant correlation with recurrence rates (p=0.004), or with RFS. Location was not found to be significant on multivariate analysis, suggesting that it is a dependent variable. Hypothalamic/midline tumors are difficult to reach and harder to remove completely or there may be additional confounding factors and our dataset may not be large enough to tease out these specific factors, so that a significance may be observed in larger studies with longer follow-up. There was no statistically significant difference in RFS between infratentorial and supratentorial tumors, with supratentorial tumors having a minimally higher recurrence rate on univariate analysis that disappeared on multivariate analysis. Histological and molecular/genetic features We identified 6 patients with PMA, and 6 other cases with histological evidence of anaplasia on pathological evaluation. The remaining 487 demonstrated histological features diagnostic of PA, occasionally aided by immunohistochemical evaluation. It was not possible to make a statistical analysis due to limited numbers of cases with unusual histological features. Likewise, a meaningful analysis of RFS for PMA and tumors with anaplastic histology could not be made. Salient features of PMAs and tumors with anaplastic histology are presented on tables 3 and 4, respectively. Histological features were critical for the recognition of PA, but their analysis did not reveal statistically meaningful associations (data not shown). Table 3. Clinical and pathological features of patients with tumors showing anaplastic histology
Table 4. Clinical and pathological features of patients with pilomyxoid astrocytoma
Because of the retrospective nature of this study and due to the long period of study, the type of molecular testing significantly varied within the group. Tumors from earlier years were tested either with Sanger sequencing for BRAF mutations or with FISH for BRAF duplication (n=191). In addition, 106 more recent cases were analyzed with a UCSF 500 NGS platform. Twenty-four of the cases had confirmed germline NF1 mutation (while NF1 mutations were detected in 5 additional tumors, the UCSF500 NGS platform was conducted solely on tumor tissue in these cases, making it impossible to determine whether these mutations were germline or somatic.) One of the tumors harbored both FGFR1 and NF1 mutations. The distribution of mutations is displayed in table 5. Table 5. Mutations in 199 pilocytic astrocytomas
Among 199 cases that had molecular analysis performed, the majority (n=150, 75.4%) had alterations in BRAF gene (either p.V600 mutations, internal tandem duplication and or KIAA1549::BRAF fusion); and the majority of the rest had alterations in other components of MAPK pathway. Because of the small number of tumors in other groups, it was not possible to make a statistical analysis between the type of mutation and RFS. We analyzed whether tumors with distinct genetic alterations cluster in particular locations. As anticipated, BRAF alterations were the most frequent type of mutations in all locations. However, BRAF alterations were more frequent in posterior fossa tumors (81%) in comparison to supratentorial tumors (69.4%; p<0.001). Notably, FGFR1 mutations were primarily observed in supratentorial tumors, with seven out of eight cases located in the hemispheric, hypothalamic-suprachiasmatic, or intraventricular regions. Although the number of cases was limited, all seven cases with mutations in other components of the MAPK pathway (KRAS, RAF1, SOS1) were supratentorial. Table 6 shows the distribution of mutations across tumor locations. Table 6. Distribution of specific mutations across tumor locations
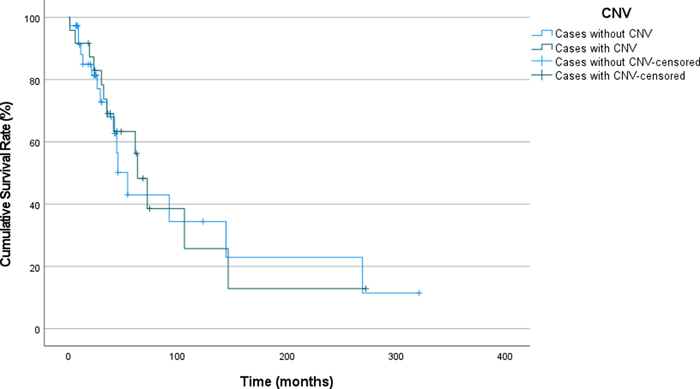
Homozygous loss of CDKN2A has been reported as one of the poor prognostic factors in the literature [44]. Among 106 patients where this information was available, CDKN2A homozygous loss occurred in only 4 patients. Histologically, all four tumors had anaplastic histologic features, typical MAPK driver mutations (NF1 mutation in two cases; FGFR1 mutation in, one case; KIAA1549::BRAF fusion in one case), additional molecular changes (ATRX mutations in three cases) and multiple chromosomal gains and losses (see Table 3). All four patients had suffered recurrences with a median time to recurrence of 35.5 months (6, 35, 35, 146 months). In 102 tumors without CDKN2A alterations (102/106), 26 of the tumors recurred with a median RFS of 32 months (1 - 269 months). Chromosomal copy number variation (CNV) analysis was done in 106 cases and showed at least one CNV in 39 tumors (36.8%). Nineteen tumors had whole chromosomal gains only, while 27 tumors had either gains or losses. In 12 tumors, partial losses or gains were recorded. Statistical analysis did not show a significant effect of the presence of copy number alteration at any degree on recurrence or RFS (p=0.816, Figure 2). There was no association between any type of genetic alteration (BRAF fusion, FGFR1 alteration, etc.) with the presence or extent of copy number loss or gain.
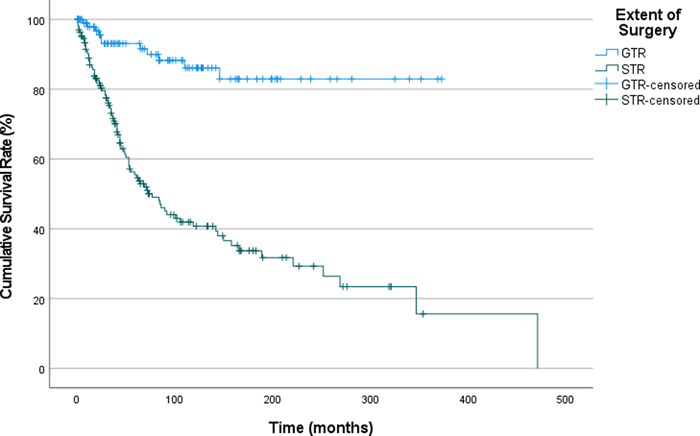
Figure 2. Recurrence-Free Survival in Patients with Pilocytic Astrocytoma, Stratified by Presence of Copy Number Variations (CNV: Copy number variation). We have also analyzed impact of chromosomal losses and gains separately. Partial and/or whole chromosomal loss was detected in 23 patients; of which, 8 was whole chromosomal loss. Partial and/or whole chromosomal gain was observed in 34 patients; of which 23 was whole chromosomal gains. Cases with any degree of chromosomal loss had a median RFS of 63 months whereas, it was 54 months for cases without any chromosomal losses. On the other hand, cases with chromosomal gain of any size had a median RFS of 63 months, whereas it was 54 months for cases without any chromosomal gains. No significant influence of whole chromosomal losses or gains was seen on recurrence, RFS and underlying molecular features of the tumor as well. Previous studies proposed whole chromosomal gains as an age-associated alteration without an impact on outcome [45]. There were 14 cases with whole chromosomal gains with no other CNV. Thus, we excluded these cases and conducted the analysis on the remaining 25. The results showed no statistically significant difference between cases with or without CNV (excluding whole chromosomal gains). Impact of age on recurrence-free survival The recurrence rates for patients younger than 14 years, between 14 and 21 years, and 21 years and over were 40.9%, 34.6%, and 20.4%, respectively. Although the recurrence rates are higher in the youngest age group, this difference was not statistically significant. (p=0.003). Among patients who underwent subtotal resection, patients older than 21 years at diagnosis showed higher recurrence rates (p<0.001), while there was no difference between age groups among patients who underwent GTR. We further investigated the confounding factors on the difference in STR group and included tumor location; even after including tumor location as a variable, patients >21 years of age showed a significantly higher recurrence rate (p<0.001). Impact of extent of resection on recurrence-free survival One-hundred-sixty-nine (33.9%) patients underwent GTR and 235 (47.1%) had STR. The data on the extent of surgery was missing in 95 (19.0%) cases. Recurrence rates were 8.5% and 20% for patients who underwent GTR and STR, respectively. The mean RFS was 321 months for patients who underwent GTR (CI: 292.3 – 350.1), and 160.9 months (CI:124.5 – 197.4) for patients who underwent STR. The patients with GTR have a significantly longer recurrence-free survival (RFS) compared to those with STR (log rank p <0.001, Figure 3). The median survival probability for overall survival could not be calculated due to absence of effect of extension of surgery on overall survival.
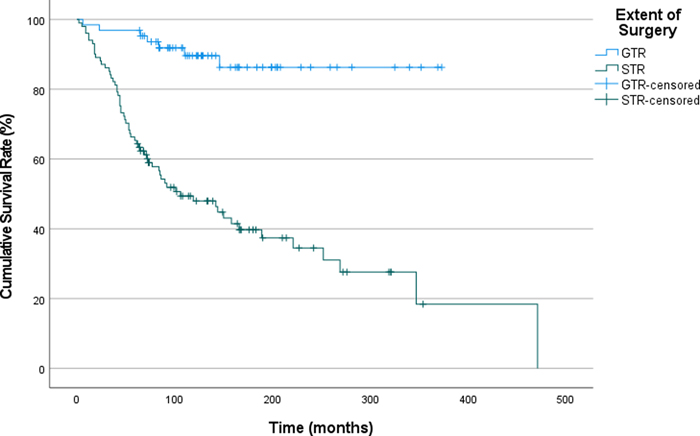
Figure 3. Recurrence-Free Survival in Patients with Pilocytic Astrocytoma, Stratified by the Extent of Surgical Resection (GTR: Gross total resection, STR: Subtotal resection). Impact of chemotherapy and/or radiotherapy on recurrence-free survival Among 81 patients who received chemotherapy, 77 had a prior STR. Patients who received chemotherapy demonstrated a shorter median RFS (141 vs 253 months). However, we think that this finding could be potentially skewed due to selection bias, as the patients with clinically more aggressive appearing tumors are more likely to receive chemotherapy. A great majority of cases (97.5%) received chemotherapy had subtotal resections, which strongly supports our hypothesis on selection bias. Thus, the observed difference in RFS may not be entirely surprising. A similar pattern was also observed among patients treated with radiotherapy (151 vs 243 months). Review of patients with neurofibromatosis 1 There were 24 confirmed neurofibromatosis Type 1 patients (11 female and 13 male), which comprise 4.8% of the entire cohort. Mean age at diagnosis was 19.5 years (3.1 – 42.7). Majority of the tumors (n=16) were in the posterior fossa, and four tumors were in the hypothalamic-suprachiasmatic region. Out of 15 patients with sufficient clinical data, three experienced recurrences. One patient died during follow-up period due to other causes (for details see table 1, case#454). Two cases had anaplastic features on histology, and the remaining 22 had classical PA morphology. None of the NF1-associated cases had mutations in other genes activating MAPK pathway (BRAF, FGFR1, etc.). Review of patients with >5-year follow-up Given PAs grow slowly and sufficiently long follow-up times are crucial for a realistic assessment of outcome, we performed a subgroup analysis of the patients who have more than 5 years of follow-up (n=178). Mean follow up time in this subgroup was 263.3 months; median follow up time was 252 months. Out of the 164 patients in this group for whom we had adequate recurrence data, 68 experienced a recurrence. The type of surgery remained one of the strongest factors that determined the rate of recurrence and RFS, as cases with STR recurred more often and earlier (Figure 4). Sixty-one percent of the patients with STR experienced recurrence at least once whereas the recurrence rate was 11% for patients with GTR (p<0.001). We were not able to show an association between the underlying genetic alterations or tumor location and recurrence rate or RFS. The number of cases in each group was not sufficient to perform an analysis of RFS for cases with and without CNV.
Figure 4. Subgroup Analysis of Recurrence-Free Survival in Patients with Pilocytic Astrocytoma Followed for >60 Months, Stratified by the Extent of Surgical Resection (GTR: Gross total resection, STR: Subtotal resection). After conducting a multivariate analysis of RFS considering the age groups, extent of resection, tumor location, NF1 status, and treatment modalities, we found that only the extent of resection had a statistically significant association with RFS (Table 7). Table 7. Multivariate analysis of factors for recurrence free survival probability
Discussion Multivariate analysis of outcome did not reveal any significant association with clinical and pathological variables except for extent of resection. Despite the large number of patients and follow-up time that is considerably longer than most studies in the literature, age, NF1 status, tumor location, adjuvant treatment and molecular alterations did not significantly influence RFS. These findings are at odds with some of the studies that suggest differences in outcome between adult and pediatric patients or between germline NF1 and wildtype tumors [46-48]. Some suggested that adult cases have a worse prognosis than pediatric counterparts [18-21] while others demonstrated a very favorable prognosis regardless of age [18, 49]. It is well recognized that older age is associated with a higher incidence of deaths compared to younger populations in patients with tumors. This is often attributed to accelerated epigenetic age and the simple effect on age on overall survival as opposed to factors directly associated with tumor progression [50, 51]. In our series, we have separated cases into three age groups and although recurrence rates increased with age, this difference was not significant. Even in subgroup analysis of cases with GTR and STR, age did not stand up as a significant factor. We were not able to assess PA specific overall survival because we observed no PA-related fatality. It has been noted that location could be an independent prognostic variable and tumors located in the posterior fossa generally have more favorable outcomes [26, 52, 53]. This was presumed to be associated with surgical access to tumor and ability to remove completely, and therefore tumors located in regions difficult to access - specifically the brain stem and hypothalamo-suprachiasmatic region - were associated with worse outcomes [20, 54]. One particular study shows that while tumor location initially appears to be a strong prognosticator, its significance diminishes when considering the extent of the resection [55]. Our analysis failed to reveal a statistically significant relationship between location and recurrence rate or RFS, despite re-categorizing cases into narrower, and later, two broad groups - "hypothalamo-suprachiasmatic + brainstem" and "other locations". The discrepancies between our findings and those in other studies may be due to the acceptance of univariate calculations as significant, even though they may not hold up on multivariate analyses when all pertinent variables, especially extent of resection, are considered. Another problem is the immediate acceptance of p<0.05 as a significant cut-off to determine biologically meaningful differences [56-58]. Yet, a series of 499 patients may still be insufficient to accurately determine the small but significant effect of some variables on outcome. One critical issue in our study is the absence of patients who died due to their disease progression and presence of rare deaths due to other factors. This could still reflect a limitation of sampling, since earlier studies reported occasional deaths due to disease progression in PA [59, 60]. The reported deaths may obviously be associated with confounding factors (such as radiation treatment or pathological misclassification) or with the assumption that the death of a patient is always a consequence of disease progression. Our data is sourced from a single referral center which minimizes certain confounding factors, allowing for a more streamlined analysis. However, the data are not associated with an epidemiologically relevant catchment area and may have selection bias due to the fact that our institution is a major national and international referral center. Therefore, our findings need further validation by large, and multi-institutional and epidemiologically relevant studies. Influence of histological features on the course of the disease has always been a debate in the literature as well as the criteria for histological anaplasia. WHO defines PA with histologic features of anaplasia as PA with brisk mitotic activity with or without necrosis. While there exist a handful of studies suggesting an association between histological anaplasia in PA and unfavorable outcome [61, 62], our findings failed to provide substantial evidence to support or refute these claims. In our series, we had only 6 cases (among 499) with histologic features of anaplasia, a sample size insufficient for meaningful comparison. These six patients were adults, with a mean age of 46.2, ranging from 27 to 72 years old. Four tumors were located in posterior fossa whereas two were hemispheric. Of these cases, five tumors were analyzed by UCSF500 NGS platform and four of them revealed a CDKN2A loss. As stated in the result section, all four cases with CDKN2A loss had histological features of anaplasia. Although there seems to be an association between CDKN2A loss and histological features of anaplasia, small number of cases prevent a meaningful and statistically significant conclusion. In our series, we identified 6 cases of PMA, which suggests that the prevalence of PMA among pilocytic tumors is only around 1%, also supported by earlier studies [29, 63]. The overall ratio and diagnostic criteria of PMA and PA with anaplastic features have been quite variable, and some published studies reported a rate of 8-20% of PMA among their PA cases [63-65]. This may reflect the challenges of making the diagnosis or the variations in histological criteria for PMA. We believe that PMA is typically a tumor of young age and hypothalamic/chiasmatic location with monomorphous histological features and deviations from this typical spectrum should be interpreted with caution [63, 65]. This could be the reason some studies did not identify significant prognostic differences between tumors designated as PMA and PA [64, 66]. Molecular studies have consistently demonstrated activation of the MAPK pathway in all cases of PA. The most prevalent genetic alteration involves kinase domain duplication and fusion of the BRAF gene with KIAA1549. Other alterations seen in PA are BRAFV600E mutations, FGFR1 alterations, NF1 alterations and also rare alterations in other components of MAPK pathway. FGFR1 alterations are highly enriched in supratentorial tumors, but spinal and cerebellar tumors also showed this molecular alteration, as observed in previous series [33, 67]. Many reports showed evidence that molecular features such as TERT promoter mutation, CDKN2A loss, TP53 mutation, or chromosomal copy number alterations may be associated with adverse clinical outcome [68-71]. In our series, we were unable to identify a meaningful association between molecular alterations and RFS. Whole or partial chromosome copy number alterations could not be correlated with outcome either. This was partially due to small numbers and partly due to short follow-up times for tumors with comprehensive molecular analysis since such analyses began only in recent years. Analysis of impact of molecular alterations including copy number variations are more likely to provide meaningful results in future studies with longer follow-up terms and larger number of patients. While molecular alterations help in establishing the diagnosis in PA, their association with location and outcome remain tenuous and require larger and prospective studies for definitive conclusions. One other point of discussion that has been a matter of contention is the decision to use the term “tumor type” instead of “tumor entity” by WHO 2021 classification. While this seems like a reasonable change prima facia due to the desire to classify tumors similar to animal and plant kingdom taxonomy, this approach entirely misses the point that tumors do not fit into neat categories of species and genera that could be easily placed in a taxonomical framework similar to animals and plants. The major objection to the use of tumor “type” is that a diagnostic entity such as PA may not be composed of a single tumor “type” but rather is a group of tumors that have sufficiently similar clinicopathological (including genetic) features to constitute a meaningful group of disorders from the perspective of the treating physician. Attempting to create “types” with every advancing bit of information would not result in the same clinically meaningful group of diseases. This is demonstrated in our study that despite their excellent outcome, tumors classified as PA harbor sufficiently different genetic alterations, radiological and pathological features that are not sufficient enough to consider them as belonging to a different entity but may arguably imply different tumor “types” or “subtypes”. In the opinion of the authors, attempting to classify tumors by placing rigid types and subtypes akin to genera, and species designations for animal and plant kingdoms may underestimate the biological diversity of pathological processes as opposed to evolutionary biology. This approach also ignores the principal necessity of trying to classify tumors to be able to manage the patients successfully. From that perspective, the tumors in this series that we believe belong to PA as an entity, have excellent long-term survival and may be considered “chronic diseases” and managed accordingly [72, 73]. Our study is in agreement with many of the prior studies highlighting the extent of resection as the key determinant of recurrence in PA [16, 53, 55, 74-76]. PA still remains a predominantly surgical disease due to the importance of extent of resection and every attempt should be made to achieve GTR for maximum benefit [77, 78]. But, there is yet much to be learned about PA, especially the biological implication of the histological features of anaplasia and additional molecular abnormalities. We were unable to identify any association between these variables and outcome, most likely because we began analyzing some of these molecular alterations only recently and sufficient time has not passed to see their effect on outcome. Prospective studies with follow up times longer than 10 years will be necessary to accurately determine the significance of molecular alterations or other variables. This will require collaborative efforts and creating tumor registries that provide platforms for longitudinal studies. References 1. P. Kleihues, F. Soylemezoglu, B. Schauble, B. W. Scheithauer, and P. C. Burger, "Histopathology, classification, and grading of gliomas", Glia, vol. 15, no. 3, pp. 211-21, Nov 1995, https://doi.org/10.1002/glia.440150303. 2. E. E. Bar, A. Lin, T. Tihan, P. C. Burger, and C. G. Eberhart, "Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma", J Neuropathol Exp Neurol, vol. 67, no. 9, pp. 878-87, Sep 2008, https://doi.org/10.1097/NEN.0b013e3181845622. 3. A. R. Elvidge, W. Penfield, and W. Cone, "The gliomas of the central nervous system. A study of 210 verified cases", Research publications - Association for Research in Nervous and Mental Disease, vol. 16, pp. 107-181, 1937. 4. H. Ohgaki and P. Kleihues, "Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas", J Neuropathol Exp Neurol, vol. 64, no. 6, pp. 479-89, Jun 2005, https://doi.org/10.1093/jnen/64.6.479. 5. C. Burkhard et al., "A population-based study of the incidence and survival rates in patients with pilocytic astrocytoma", J Neurosurg, vol. 98, no. 6, pp. 1170-4, Jun 2003, https://doi.org/10.3171/jns.2003.98.6.1170. 6. Q. T. Ostrom et al., "CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007-2011", Neuro Oncol, vol. 16 Suppl 4, no. Suppl 4, pp. iv1-63, Oct 2014, https://doi.org/10.1093/neuonc/nou223. 7. R. J. Packer et al., "Pediatric low-grade gliomas: implications of the biologic era", Neuro Oncol, vol. 19, no. 6, pp. 750-761, Jun 1 2017, https://doi.org/10.1093/neuonc/now209. 8. I. Fried, U. Tabori, T. Tihan, A. Reginald, and E. Bouffet, "Optic pathway gliomas: a review", CNS Oncol, vol. 2, no. 2, pp. 143-59, Mar 2013, https://doi.org/10.2217/cns.12.47. 9. Q. T. Ostrom et al., "CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012-2016", Neuro Oncol, vol. 21, no. Suppl 5, pp. v1-v100, Nov 1 2019, https://doi.org/10.1093/neuonc/noz150. 10. P. D. Brown et al., "Adult patients with supratentorial pilocytic astrocytoma: long-term follow-up of prospective multicenter clinical trial NCCTG-867251 (Alliance)", Neurooncol Pract, vol. 2, no. 4, pp. 199-204, Dec 2015, https://doi.org/10.1093/nop/npv031. 11. P. D. Brown et al., "Adult patients with supratentorial pilocytic astrocytomas: a prospective multicenter clinical trial", Int J Radiat Oncol Biol Phys, vol. 58, no. 4, pp. 1153-60, Mar 15 2004, https://doi.org/10.1016/j.ijrobp.2003.09.020. 12. Q. T. Ostrom, N. Patil, G. Cioffi, K. Waite, C. Kruchko, and J. S. Barnholtz-Sloan, "CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013-2017", Neuro Oncol, vol. 22, no. 12 Suppl 2, pp. iv1-iv96, Oct 30 2020, https://doi.org/10.1093/neuonc/noaa200. 13. Q. T. Ostrom, G. Cioffi, K. Waite, C. Kruchko, and J. S. Barnholtz-Sloan, "CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014-2018", Neuro Oncol, vol. 23, no. 12 Suppl 2, pp. iii1-iii105, Oct 5 2021, https://doi.org/10.1093/neuonc/noab200. 14. J. S. Nix, J. Blakeley, and F. J. Rodriguez, "An update on the central nervous system manifestations of neurofibromatosis type 1", Acta Neuropathol, vol. 139, no. 4, pp. 625-641, Apr 2020, https://doi.org/10.1007/s00401-019-02002-2. 15. C. Fernandez et al., "Pilocytic astrocytomas in children: prognostic factors--a retrospective study of 80 cases", Neurosurgery, vol. 53, no. 3, pp. 544-53; discussion 554-5, Sep 2003, https://doi.org/10.1227/01.neu.0000079330.01541.6e. 16. C. Jungk et al., "Extent of Resection, MGMT Promoter Methylation Status and Tumor Location Independently Predict Progression-Free Survival in Adult Sporadic Pilocytic Astrocytoma", Cancers (Basel), vol. 11, no. 8, Jul 29 2019, https://doi.org/10.3390/cancers11081072. 17. C. Colin et al., "Outcome analysis of childhood pilocytic astrocytomas: a retrospective study of 148 cases at a single institution", Neuropathol Appl Neurobiol, vol. 39, no. 6, pp. 693-705, Oct 2013, https://doi.org/10.1111/nan.12013. 18. Y. Tomita et al., "Age is a major determinant for poor prognosis in patients with pilocytic astrocytoma: a SEER population study", Clin Exp Med, 2023 Oct;23(6):2301-2309., https://doi.org/10.1007/s10238-022-00882-5. 19. K. J. Lee et al., "Management and Survival of Adult Patients with Pilocytic Astrocytoma in the National Cancer Database", World Neurosurg, vol. 112, pp. e881-e887, Apr 2018, https://doi.org/10.1016/j.wneu.2018.01.208. 20. W. Yang, J. L. Porras, A. M. Khalafallah, Y. Sun, A. Bettegowda, and D. Mukherjee, "Comparison of adult and pediatric pilocytic astrocytomas using competing risk analysis: A population-based study", Clin Neurol Neurosurg, vol. 212, p. 107084, Jan 2022, https://doi.org/10.1016/j.clineuro.2021.107084. 21. I. Shin et al., "Clinical factors and conventional MRI may independently predict progression-free survival and overall survival in adult pilocytic astrocytomas", Neuroradiology, vol. 64, no. 8, pp. 1529-1537, Aug 2022, https://doi.org/10.1007/s00234-021-02872-y. 22. K. J. Coakley, J. Huston, 3rd, B. W. Scheithauer, G. Forbes, and P. J. Kelly, "Pilocytic astrocytomas: well-demarcated magnetic resonance appearance despite frequent infiltration histologically", Mayo Clin Proc, vol. 70, no. 8, pp. 747-51, Aug 1995, https://doi.org/10.4065/70.8.747. 23. A. Palpan Flores, V. Rodriguez Dominguez, I. Esteban Rodriguez, M. Roman de Aragon, and A. Zamarron Perez, "H3K27M-mutant glioma in thoracic spinal cord and conus medullaris with pilocytic astrocytoma morphology: case report and review of the literature", Br J Neurosurg, pp. 1-7, Oct 7 2021, https://doi.org/10.1080/02688697.2021.1988054. 24. D. Brat et al., WHO Classification of Tumours of the Central Nervous System, 5th ed. (WHO Classification of Tumours, no. 6). Lyon: International Agency for Research on Cancer, 2021. 25. F. J. Rodriguez, B. W. Scheithauer, P. C. Burger, S. Jenkins, and C. Giannini, "Anaplasia in pilocytic astrocytoma predicts aggressive behavior", Am J Surg Pathol, vol. 34, no. 2, pp. 147-60, Feb 2010, https://doi.org/10.1097/PAS.0b013e3181c75238. 26. P. A. Forsyth, E. G. Shaw, B. W. Scheithauer, J. R. O'Fallon, D. D. Layton, Jr., and J. A. Katzmann, "Supratentorial pilocytic astrocytomas. A clinicopathologic, prognostic, and flow cytometric study of 51 patients", Cancer, vol. 72, no. 4, pp. 1335-42, Aug 15 1993, https://doi.org/10.1002/1097-0142(19930815)72:4<1335::aid-cncr2820720431>3.0.co;2-e. 27. A. Wade, C. Hayhurst, A. Amato-Watkins, A. Lammie, and P. Leach, "Cerebellar pilocytic astrocytoma in adults: a management paradigm for a rare tumour", Acta Neurochir (Wien), vol. 155, no. 8, pp. 1431-5, Aug 2013, https://doi.org/10.1007/s00701-013-1790-1. 28. A. Paixao Becker, R. S. de Oliveira, F. P. Saggioro, L. Neder, L. M. Chimelli, and H. R. Machado, "In pursuit of prognostic factors in children with pilocytic astrocytomas", Childs Nerv Syst, vol. 26, no. 1, pp. 19-28, Jan 2010, https://doi.org/10.1007/s00381-009-0990-8. 29. R. J. Komotar et al., "Pilocytic and pilomyxoid hypothalamic/chiasmatic astrocytomas", Neurosurgery, vol. 54, no. 1, pp. 72-9; discussion 79-80, Jan 2004, https://doi.org/10.1227/01.neu.0000097266.89676.25. 30. E. T. Hidalgo et al., "Long-term clinical and visual outcomes after surgical resection of pediatric pilocytic/pilomyxoid optic pathway gliomas", J Neurosurg Pediatr, vol. 24, no. 2, pp. 166-173, May 17 2019, https://doi.org/10.3171/2019.2.PEDS18529. 31. R. Parisi et al., "Multi-institution analysis of tumor mutational burden and outcomes in pediatric central nervous system tumor patients", Pediatr Blood Cancer, vol. 70, no. 3, p. e30139, Mar 2023, https://doi.org/10.1002/pbc.30139. 32. V. Thatikonda et al., "Comprehensive analysis of mutational signatures reveals distinct patterns and molecular processes across 27 pediatric cancers", Nat Cancer, vol. 4, no. 2, pp. 276-289, Feb 2023, https://doi.org/10.1038/s43018-022-00509-4. 33. D. T. Jones et al., "Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma", Nat Genet, vol. 45, no. 8, pp. 927-32, Aug 2013, https://doi.org/10.1038/ng.2682. 34. J. Zhang et al., "Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas", Nat Genet, vol. 45, no. 6, pp. 602-12, Jun 2013, https://doi.org/10.1038/ng.2611. 35. K. Jacob et al., "Duplication of 7q34 is specific to juvenile pilocytic astrocytomas and a hallmark of cerebellar and optic pathway tumours", Br J Cancer, vol. 101, no. 4, pp. 722-33, Aug 18 2009, https://doi.org/10.1038/sj.bjc.6605179. 36. T. Forshew et al., "Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas", J Pathol, vol. 218, no. 2, pp. 172-81, Jun 2009, https://doi.org/10.1002/path.2558. 37. D. Salles, G. Laviola, A. C. M. Malinverni, and J. N. Stavale, "Pilocytic Astrocytoma: A Review of General, Clinical, and Molecular Characteristics", J Child Neurol, vol. 35, no. 12, pp. 852-858, Oct 2020, https://doi.org/10.1177/0883073820937225. 38. Y. H. Chen and D. H. Gutmann, "The molecular and cell biology of pediatric low-grade gliomas", Oncogene, vol. 33, no. 16, pp. 2019-26, Apr 17 2014, https://doi.org/10.1038/onc.2013.148. 39. T. T. Tomic et al., "A new GTF2I-BRAF fusion mediating MAPK pathway activation in pilocytic astrocytoma", PLoS One, vol. 12, no. 4, p. e0175638, 2017, https://doi.org/10.1371/journal.pone.0175638. 40. A. P. Becker et al., "KIAA1549: BRAF Gene Fusion and FGFR1 Hotspot Mutations Are Prognostic Factors in Pilocytic Astrocytomas", J Neuropathol Exp Neurol, vol. 74, no. 7, pp. 743-54, Jul 2015, https://doi.org/10.1097/NEN.0000000000000213. 41. C. Stuer, B. Vilz, M. Majores, A. Becker, J. Schramm, and M. Simon, "Frequent recurrence and progression in pilocytic astrocytoma in adults", Cancer, vol. 110, no. 12, pp. 2799-808, Dec 15 2007, https://doi.org/10.1002/cncr.23148. 42. D. J. Cote et al., "Glioma incidence and survival variations by county-level socioeconomic measures", Cancer, vol. 125, no. 19, pp. 3390-3400, Oct 1 2019, https://doi.org/10.1002/cncr.32328. 43. C. N. Kline et al., "Targeted next-generation sequencing of pediatric neuro-oncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy", Neuro Oncol, vol. 19, no. 5, pp. 699-709, May 1 2017, https://doi.org/10.1093/neuonc/now254. 44. A. Reinhardt et al., "Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations", Acta Neuropathol, vol. 136, no. 2, pp. 273-291, Aug 2018, https://doi.org/10.1007/s00401-018-1837-8. 45. K. Chatsirisupachai, T. Lesluyes, L. Paraoan, P. Van Loo, and J. P. de Magalhaes, "An integrative analysis of the age-associated multi-omic landscape across cancers", Nat Commun, vol. 12, no. 1, p. 2345, Apr 20 2021, https://doi.org/10.1038/s41467-021-22560-y. 46. R. E. Strowd, 3rd et al., "Histologically benign, clinically aggressive: Progressive non-optic pathway pilocytic astrocytomas in adults with NF1", Am J Med Genet A, vol. 170, no. 6, pp. 1455-61, Jun 2016, https://doi.org/10.1002/ajmg.a.37622. 47. J. Helfferich et al., "Neurofibromatosis type 1 associated low grade gliomas: A comparison with sporadic low grade gliomas", Crit Rev Oncol Hematol, vol. 104, pp. 30-41, Aug 2016, https://doi.org/10.1016/j.critrevonc.2016.05.008. 48. V. Laithier et al., "Progression-free survival in children with optic pathway tumors: dependence on age and the quality of the response to chemotherapy--results of the first French prospective study for the French Society of Pediatric Oncology", J Clin Oncol, vol. 21, no. 24, pp. 4572-8, Dec 15 2003, https://doi.org/10.1200/JCO.2003.03.043. 49. D. R. Johnson, P. D. Brown, E. Galanis, and J. E. Hammack, "Pilocytic astrocytoma survival in adults: analysis of the Surveillance, Epidemiology, and End Results Program of the National Cancer Institute", J Neurooncol, vol. 108, no. 1, pp. 187-93, May 2012, https://doi.org/10.1007/s11060-012-0829-0. 50. S. Shafqat, E. Arana Chicas, A. Shafqat, and S. K. Hashmi, "The Achilles' heel of cancer survivors: fundamentals of accelerated cellular senescence", J Clin Invest, vol. 132, no. 13, Jul 1 2022, https://doi.org/10.1172/JCI158452. 51. A. M. Molinaro et al., "Interactions of Age and Blood Immune Factors and Noninvasive Prediction of Glioma Survival", J Natl Cancer Inst, vol. 114, no. 3, pp. 446-457, Mar 8 2022, https://doi.org/10.1093/jnci/djab195. 52. S. Cyrine et al., "Pilocytic astrocytoma: a retrospective study of 32 cases", Clin Neurol Neurosurg, vol. 115, no. 8, pp. 1220-5, Aug 2013, https://doi.org/10.1016/j.clineuro.2012.11.009. 53. T. Kayama, T. Tominaga, and T. Yoshimoto, "Management of pilocytic astrocytoma", Neurosurg Rev, vol. 19, no. 4, pp. 217-20, 1996, https://doi.org/10.1007/BF00314833. 54. J. M. Ye, M. J. Ye, S. Kranz, and P. Lo, "A 10 year retrospective study of surgical outcomes of adult intracranial pilocytic astrocytoma", J Clin Neurosci, vol. 21, no. 12, pp. 2160-4, Dec 2014, https://doi.org/10.1016/j.jocn.2014.04.015. 55. J. W. Kim et al., "Comparison of the clinical features and treatment outcomes of pilocytic astrocytoma in pediatric and adult patients", Childs Nerv Syst, vol. 39, no. 3, pp. 583-591, Mar 2023, https://doi.org/10.1007/s00381-023-05839-x. 56. R. Jimenez-Paneque, "The questioned p value: clinical, practical and statistical significance", Medwave, vol. 16, no. 8, p. e6534, Sep 9 2016, https://doi.org/10.5867/medwave.2016.08.6534. El valor de p en entredicho: significacion estadistica, clinica y practica. 57. P. R. Burton, L. C. Gurrin, and M. J. Campbell, "Clinical significance not statistical significance: a simple Bayesian alternative to p values", J Epidemiol Community Health, vol. 52, no. 5, pp. 318-23, May 1998, https://doi.org/10.1136/jech.52.5.318. 58. P. Martinez-Camblor, S. Perez-Fernandez, and S. Diaz-Coto, "The role of the p-value in the multitesting problem", J Appl Stat, vol. 47, no. 9, pp. 1529-1542, 2020, https://doi.org/10.1080/02664763.2019.1682128. 59. J. Coelho, S. Nunes, and D. Salgado, "Spontaneous Malignant Transformation of a Pilocytic Astrocytoma of Cerebellum: Case Report", Child Neurol Open, vol. 2, no. 1, p. 2329048X14566813, Jan-Mar 2015, https://doi.org/10.1177/2329048X14566813. 60. J. A. Ellis, A. Waziri, C. Balmaceda, P. Canoll, J. N. Bruce, and M. B. Sisti, "Rapid recurrence and malignant transformation of pilocytic astrocytoma in adult patients", J Neurooncol, vol. 95, no. 3, pp. 377-382, Dec 2009, https://doi.org/10.1007/s11060-009-9935-z. 61. C. Dix et al., "C-reactive protein, immunothrombosis and venous thromboembolism", Front Immunol, vol. 13, p. 1002652, 2022, https://doi.org/10.3389/fimmu.2022.1002652. 62. F. J. Rodriguez et al., "Alternative lengthening of telomeres, ATRX loss and H3-K27M mutations in histologically defined pilocytic astrocytoma with anaplasia", Brain Pathol, vol. 29, no. 1, pp. 126-140, Jan 2019, https://doi.org/10.1111/bpa.12646. 63. T. Tihan et al., "Pediatric astrocytomas with monomorphous pilomyxoid features and a less favorable outcome", J Neuropathol Exp Neurol, vol. 58, no. 10, pp. 1061-8, Oct 1999, https://doi.org/10.1097/00005072-199910000-00004. 64. D. Bhargava et al., "Occurrence and distribution of pilomyxoid astrocytoma", Br J Neurosurg, vol. 27, no. 4, pp. 413-8, Aug 2013, https://doi.org/10.3109/02688697.2012.752430. 65. K. Chikai et al., "Clinico-pathological features of pilomyxoid astrocytoma of the optic pathway", Acta Neuropathol, vol. 108, no. 2, pp. 109-14, Aug 2004, https://doi.org/10.1007/s00401-004-0858-7. 66. E. AlShail et al., "A molecular study of pediatric pilomyxoid and pilocytic astrocytomas: Genome-wide copy number screening, retrospective analysis of clinicopathological features and long-term clinical outcome", Front Oncol, vol. 13, p. 1034292, 2023, https://doi.org/10.3389/fonc.2023.1034292. 67. C. G. Lucas et al., "Comprehensive analysis of diverse low-grade neuroepithelial tumors with FGFR1 alterations reveals a distinct molecular signature of rosette-forming glioneuronal tumor", Acta Neuropathol Commun, vol. 8, no. 1, p. 151, Aug 28 2020, https://doi.org/10.1186/s40478-020-01027-z. 68. R. Tamura et al., "Clinical, histopathological and molecular risk factors for recurrence of pilocytic astrocytomas: brainstem/spinal location, nestin expression and gain of 7q and 19 are associated with early tumor recurrence", Brain Tumor Pathol, vol. 40, no. 2, pp. 109-123, Apr 2023, https://doi.org/10.1007/s10014-023-00453-w. 69. J. V. Francis, B. Tanrikulu, A. E. Danyeli, and M. M. Ozek, "Is it a new culprit? "TERT promoter mutation" in an aggressive pediatric pilocytic astrocytoma", Childs Nerv Syst, vol. 37, no. 3, pp. 1003-1008, Mar 2021, https://doi.org/10.1007/s00381-020-04803-3. 70. D. N. Cagney et al., "Clinical Importance of CDKN2A Loss and Monosomy 10 in Pilocytic Astrocytoma", Cureus, vol. 11, no. 5, p. e4726, May 23 2019, https://doi.org/10.7759/cureus.4726. 71. E. F. Rodriguez et al., "PI3K/AKT pathway alterations are associated with clinically aggressive and histologically anaplastic subsets of pilocytic astrocytoma", Acta Neuropathol, vol. 121, no. 3, pp. 407-20, Mar 2011, https://doi.org/10.1007/s00401-010-0784-9. 72. G. F. Reis and T. Tihan, "Practical molecular pathologic diagnosis of pilocytic astrocytomas", Surg Pathol Clin, vol. 8, no. 1, pp. 63-71, Mar 2015, https://doi.org/10.1016/j.path.2014.10.002. 73. P. Daszkiewicz, A. Maryniak, M. Roszkowski, and S. Barszcz, "Long-term functional outcome of surgical treatment of juvenile pilocytic astrocytoma of the cerebellum in children", Childs Nerv Syst, vol. 25, no. 7, pp. 855-60, Jul 2009, https://doi.org/10.1007/s00381-009-0855-1. 74. K. M. Bond, J. D. Hughes, A. L. Porter, J. Orina, S. Fang, and I. F. Parney, "Adult Pilocytic Astrocytoma: An Institutional Series and Systematic Literature Review for Extent of Resection and Recurrence", World Neurosurg, vol. 110, pp. 276-283, Feb 2018, https://doi.org/10.1016/j.wneu.2017.11.102. 75. A. N. Santos et al., "Long-term outcome after management of pilocytic astrocytoma in the posterior fossa in a pediatric population", IBRO Neurosci Rep, vol. 13, pp. 388-392, Dec 2022, https://doi.org/10.1016/j.ibneur.2022.10.001. 76. B. A. Muhsen, A. I. Aljariri, M. Elayyan, H. Hirbawi, and M. A. Masri, "Insight about the characteristics and surgical resectability of adult pilocytic astrocytoma: tertiary center experience", CNS Oncol, vol. 11, no. 1, p. CNS81, Apr 6 2022, https://doi.org/10.2217/cns-2021-0014. 77. L. Y. Wen et al., "Associations between Chinese college students' anxiety and depression: A chain mediation analysis", PLoS One, vol. 17, no. 6, p. e0268773, 2022, https://doi.org/10.1371/journal.pone.0268773. 78. S. J. Cler et al., "Genetic and histopathological associations with outcome in pediatric pilocytic astrocytoma", J Neurosurg Pediatr, vol. 29, no. 5, pp. 504-512, May 1 2022, https://doi.org/10.3171/2021.9.PEDS21405.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||