|
|
|
Free Neuropathology 4:14 (2023) |
|
Review |
|
Neurotrauma: 2023 Update |
|
David S. Priemer1-3, Daniel P. Perl1,2 |
|
|
Corresponding author: |
|
Submitted: 18 August 2023 |
|
Keywords: Traumatic brain injury, Chronic traumatic encephalopathy, Tau, TDP-43, APOE |
|
Abstract 2022 was a productive year for research in traumatic brain injury (TBI) and resultant neuropathology. After an extensive review, we present related studies and publications which we felt were of particular importance to the neuropathology community. First, 2022 was highlighted by important advancements in the diagnosis and, moreover, our understanding of chronic traumatic encephalopathy (CTE). Important publications include a pair concluding that CTE primarily concerns neuronal accumulation of phosphorylated tau (ptau), but that glial ptau accumulation often helps to facilitate diagnosis. In addition, a new large community study from Australia continues the indication that CTE is relatively uncommon in the community, and the first large-cohort study on brains of military personnel similarly demonstrates that CTE appears to be uncommon among service members and does not appear to explain high rates of neuropsychiatric sequelae suffered by the warfighter. The causation of CTE by impact-type TBI was supported by the application of the Bradford Hill criteria, within the brains of headbutting bovids, and interestingly within an artificial head model exposed to linear impact. Finally, a large-scale analysis of APOE genotypes contends that gene status may influence CTE pathology and outcomes. In experimental animal work, a study using mouse models provided important evidence that TDP-43 facilitates neurodegenerative pathology and is implicated in cognitive dysfunction following TBI, and another study using a swine model for concussion demonstrated that evidence that axonal sodium channel disruption may be a driver of neurologic dysfunction after concussion. Finally, we end with memoriam to Dr. John Q. Trojanowski, a giant of neurodegenerative research and an important contributor to the neurotrauma literature, who we lost in 2022. |
|
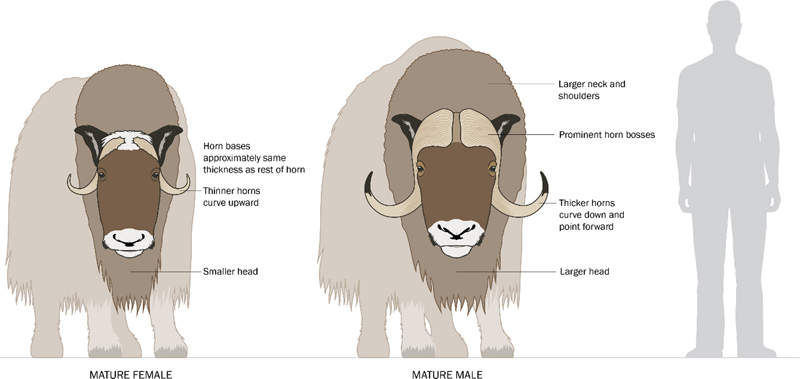
Introduction The year 2022 was an exciting and highly productive year for research in traumatic brain injury (TBI). Accordingly, a large number of papers with notable impact to the neuropathology community were published, and the task of highlighting only a small handful of them for this review proved extraordinarily difficult. Nonetheless, and after a thorough review of the literature, herein we recognize the papers that what we felt were of particular interest to the neuropathology community, with a summary of their contribution to our field and thus why we recognized them. As you will see, many of our selections this year involve important work within the field of chronic traumatic encephalopathy (CTE). We readily acknowledge that inclusions and exclusions from this list reflect our own bias, and that some of the papers we selected involve our own research efforts. We suspect that at least several of our considerations would have also been chosen by others if given the same task. Finally, we conclude our review with memoriam to the life and exemplary career of Dr. John Q. Trojanowski, whose unexpected death in 2022 left a legacy that will not soon be forgotten. Chronic traumatic encephalopathy is caused by repetitive impact traumatic brain injury In 1965, Sir Austin Bradford Hill proposed what became known as the Bradford Hill Criteria. The Bradford Hill Criteria provide a system to examine the potential role of environmental exposures in causation of disease.1 Bradford Hill proposed the use of nine different criteria or “viewpoints”, and these were primarily aimed at establishing a causal relationship between smoking and lung cancer. In 2022, authors Nowinski et al. attempted to apply all nine viewpoints of the Bradford Hill criteria through a review of literature in order to investigate the relationship between repetitive TBI and subsequent CTE pathology.2 What they established in their review was that: CTE pathology had considerable strength of association with repetitive TBI (Bradford Hill criterion 1); the relationship between repetitive TBI and CTE has been consistently demonstrated across multiple studies conducted by independent groups in different patient populations (criterion 2); there is evidence of a dose-response relationship between amount of repetitive TBI over time and CTE (e.g., association between years played in contact sports and increasing likelihood to have CTE at autopsy; criterion 5); there is evidence suggesting mechanistic plausibility that TBI can result in the unique distribution of phosphorylated tau (ptau) pathology in CTE (criterion 6); an association between repetitive TBI and CTE is not in conflict with what we know about both TBI and CTE (i.e., coherence, criterion 7); and the relationship between repetitive TBI and CTE is analogous to similar causal associations (criterion 9). Criticism of the application of the Bradford Hill Criteria to repetitive TBI and CTE may emerge particularly on the topics of specificity (criterion 3) and temporality (criterion 4), as the literature indicates that CTE is not entirely specific to repetitive TBI3-5, and it is difficult to definitively establish a temporal relationship with repetitive TBI and CTE in a situation where the disease can only be diagnosed post mortem. Finally, the 8th criterion examining experimental evidence demonstrating causality from an environmental exposure, or demonstrating prevention of disease from cessation of an environmental exposure, has not yet been fulfilled in regard to CTE and repetitive TBI, and may take many years to definitively address. However, regardless of whether all nine criteria are met, the application of this method to examine the relationship between repetitive TBI and CTE is commendable and has made an important contribution to the literature. Chronic traumatic encephalopathy-like pathology in the brains of headbutting animals In support of the relationship between repetitive TBI and CTE, an interesting study also emerged in 2022 from a unique and unusual source: animals who engage in headbutting. Authors Ackermans et al. performed postmortem neuropathologic examination of brains obtained from muskoxen and bighorn sheep, two animal species that are well known to engage in headbutting displays at high speeds and with large applied forces.6 Brains from seven animals were included, (four bighorn sheep, three muskoxen), involving both sexes and a distribution of ages from young-to-middle age in bighorn sheep and middle-to-old age in the muskoxen. All three muskoxen were from the wild, while three of the four bighorn sheep were from captivity. Examination was of the prefrontal cortex +/- the parietal cortex and used three different antibodies for ptau (CP13, AT8, and PHF-1) among other markers. The authors found high degrees ptau immunoreactivity in neurons/neuronal structures in all muskoxen samples, often preferentially in distributions reminiscent of CTE (with preference at the bottom of sulci, occasionally in a perivascular distribution, and in superficial cortical layers), and to lesser degrees in the brain of the single male bighorn sheep examined. The pathology was observed in the absence of Aβ plaques. The authors speculate that the reduced frequency of ptau pathology in bighorn sheep may be due to the relatively young age and/or captive status of the animals. Interestingly, and even though male muskoxen headbutt more frequently and with greater force than female, the female muskoxen tended to demonstrate more severe ptau pathology than the male. The authors suggest that this may be due to individual differences (small sample size), or anatomical differences in the male such as increased skull thickness and more pronounced forehead fat pads (Figure 1). Regardless of these details, this study provides the first direct evidence of ptau accumulation in patterns similar to CTE within the brains gyrencephalic animals naturally exposed to repetitive TBI. Further investigation of brains from additional animals whose nature exposes them to repetitive TBI is warranted, and encouraged.
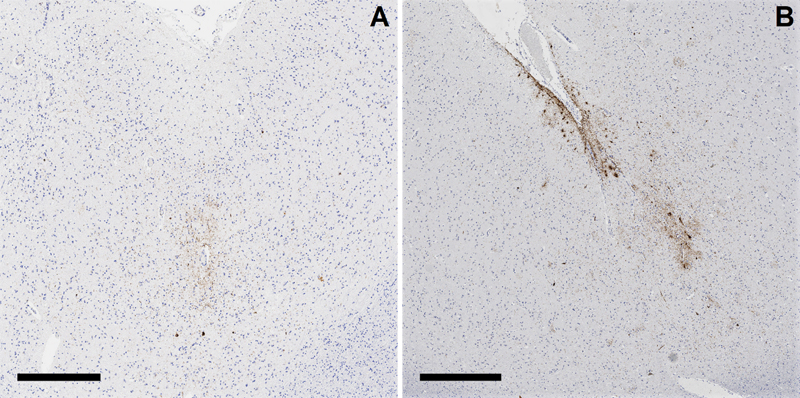
Figure 1. Comparison of male and female muskoxen. Anatomical differences in male versus female oxen may play a role in protecting male muskoxen from traumatic brain injury relative to females. Mechanistic evidence for sulcal depth-predominant injury from impact brain trauma Mechanistic plausibility for the distinct pattern of sulcal depth pathology in CTE as due to impact TBI was further supported in 2022, with an important work from researchers Kerwin et al. titled “Sulcal Cavitation in Linear Head Acceleration: Possible Correlation with Chronic Traumatic Encephalopathy”.7 The researchers applied linear impact, via a drop from height, to an artificial, polymer-based head model (“Biofidelic Brain Phantom”) complete with a 3D-printed skull, deionized water representing cerebrospinal fluid, and a simplified cerebrum constructed with grey and white matter components at appropriately different weight concentrations (i.e., densities), and with gyri and sulci. The model was designed to create a force of impact similar to an average boxing punch, and the physical distortion of the artificial brain to the linear impact was recorded at 25,000 frames/second. Researchers qualitatively observed a pattern of cavitation within the brain, via the formation of vapor bubbles (“inertial cavitation bubbles”) due to the change in intracranial pressure induced by the impact, most notably in the contrecoup region. Fascinatingly, their subsequent computational measurements showed that the strain from the fluid cavitation was particularly focused at depths of sulci, providing evidence that physical disruption to the brain from impact head injury is most pronounced at depths of sulci and thus consistent with the pattern of pathology observed in CTE. Importantly, in the year prior the same laboratory published the results from a different experiment in which the artificial brain model was exposed to blast wave; this resulted in an entirely different pattern of tissue distortion with physical strain maximized at interface regions, particularly the subpial region and gray-white matter junction, closely corresponding to the pattern of pathology observed in interface astroglial scarring.8,9 Chronic traumatic encephalopathy is primarily a neuronal disease Debate over how to identify and classify CTE pathology continued in 2022, with a focus being on the relevance of astrocytic ptau pathology in cases of CTE and largely in response to the de-emphasis of astrocytic ptau as necessary for a CTE diagnosis in the most recent consensus diagnostic statement.10 Authors Butler et al., with the resources of the Boston University CTE Brain Bank, examined dorsolateral frontal cortex samples from the brains of 150 decedents with known CTE for the precise cellular distribution of ptau pathology in the cerebral cortex.11 Individuals whose brains were examined in the study ranged from 21 to 80 years of age. Analysis of which cell types were involved by ptau pathology was accomplished via co-labeling with markers for astrocytes (GFAP) and neurons (MAP2). The authors identified that not only was neuronal ptau predominant in cases of CTE, but extent of neuronal ptau pathology was significantly correlated with age, extent/years of repetitive TBI exposure, and assessed severity of CTE, while astrocytic ptau pathology only had a significant association with age. Indeed, many brains from younger decedents (age of death <40) with diagnostic CTE pathology had sparse, if any, astrocytic ptau pathology (Figure 2A). The authors also observed that at sulcal depths glial ptau pathology was largely superficial/subpial in orientation, whereas neurons were predominant in the distinctive perivascular localization of ptau pathology of CTE (Figure 2B). While they could not refute that astrocytic ptau pathology may be exacerbated by CTE, particularly at sulcal depths, or that astrocytic ptau pathology may play some role in CTE pathobiology, the authors ultimately conclude that astrocytic ptau pathology in CTE is an age-driven finding akin to age-related tau astrogliopathy (ARTAG), which itself is also not considered to be related to TBI exposure. As such, astrocytic ptau in CTE cases may simply represent a co-existent pathology in brains from aged individuals.
Figure 2. Chronic traumatic encephalopathy (CTE) pathognomonic lesions without (A) and with (B) superficial glial phosphorylated ptau accumulation. Evidence from 2022 supports that CTE is primarily a neuronal disease, in that neuronal ptau pathology in CTE cases is significantly correlated with age, extent/years of repetitive TBI exposure, and severity of CTE, while astrocytic ptau pathology is only correlated with age. However, other research also suggests that astrocytic accumulation of ptau may nonetheless facilitate better recognition of a pathognomonic CTE lesion in practice. (Scale bars: A, 500µm; B, 700µm) Astrocytic ptau pathology aids the recognition of chronic traumatic encephalopathy pathology Although astrocytic ptau in the context of CTE seems to be age-driven and possibly independent of TBI exposure, the presence of astrocytic ptau pathology, or at least ensuring that the antibodies used to identify ptau include astrocytic ptau, may nonetheless be critical to enabling the reliable diagnosis of CTE in clinical practice. Authors Ameen-Ali et al., including the investigators of the CONNECT-TBI research group, published fascinating data in this regard within their manuscript entitled “Detection of astrocytic tau pathology facilitates recognition of [CTE] neuropathologic change”.12 Twelve cases from contact sports athletes with known CTE were identified, along with four cases of Alzheimer disease neuropathologic change (ADNC). One cortical tissue block known to contain representative pathology was selected from each case, and four adjacent/consecutive sections from each block were stained with four different ptau antibodies, including ptau antibodies that would exclusively identify neuronal ptau in CTE (3R and GT-38) and antibodies that would label both glial and neuronal accumulations (4R, PHF-1). A total of 9 neuropathologists assessed digital slide sets that collectively included all stained sections from all sixteen cases, and did so while completely blind to case information (e.g. decedent demographics, prior neuropathologic diagnoses, etc.) and also to the precise antibody that they were evaluating on any given slide. Reviewing pathologists were then simply asked if they would classify a given slide as containing CTE pathology according to current consensus criteria, and to provide a degree of confidence in their opinion from 0-100%. The resulting data showed that the type of antibody used dramatically impacted the detection, and confidence of detection, of CTE pathology. The authors calculated significantly reduced sensitivities for the detection of CTE using antibodies that exclusively labeled neuronal ptau (GT-38, 51.9%; 3R, 15.7%) in comparison to the antibodies that were inclusive of both neuronal and astrocytic ptau (PHF-1, 80.6%; 4R, 73.0%); CTE was detected by the majority of pathologists in 75% of cases using PHF-1, 70% of cases using 4R, 57% of cases using GT-38, and in only 5% of cases using 3R. Using this novel approach, the authors conclude that identification of CTE pathology is optimized when the presence of both neuronal and astrocytic ptau accumulations can be evaluated, and the visualization of neuronal ptau pathology alone leads to inconsistent diagnoses. The juxtaposition of the data from this study and the study recognized in the prior section clearly demonstrates the ongoing need for continued refinement, validation, and investigation into the practical application of current diagnostic criteria for CTE. It is our hope that manuscripts like these will spur further critical investigations of the current CTE diagnostic criteria, and eventually the convening of additional consensus efforts. More evidence that chronic traumatic encephalopathy is uncommon in the community Continuing from important work that has suggested that CTE in the general community is rather uncommon13-15, particularly outside the context of contact sports or other forms of impact-type TBI, the year 2022 also saw a new large-cohort community-based study, this time in Australia. Researchers Suter et al. published data on prospective CTE screening of 180 consecutive brains from a routine neuropathology service, including 162 cases comprising an “unselected clinical cohort” and 18 cases which were donated due to known dementia.16 The cohort demographics were broad: 58% of the cases were from males and 42% from females, the age of death ranged from 18-101 years (average age of death: 55 years), and 14% of the cohort had known TBI exposure including from contact sports, physical assaults, neurosurgery, falls, and other sources. In addition, 28% had a history of alcohol or other substance abuse. All cases were screened for CTE as part of the initial diagnostic evaluation. For the 162 routine brain referrals the screening process included performance of ptau immunohistochemistry (AT8 antibody) on sections from three blocks containing superior and middle frontal gyri, peri-Rolandic cortex, and superior and middle temporal gyri. The known dementia donations were more extensively examined for ptau pathology in accordance with institutional dementia protocols. The researchers identified a myriad of neurodegenerative pathology in the series, including four cases (2.5%) with CTE pathology. All four CTE cases were identified following the three-block screening protocol instituted for unselected clinical cases (not donated for known dementia). All four CTE cases involved death in the sixth decade of life. Two of the four cases had known TBI history, in the form of a history of TBI from boxing and history of prior brain surgery. Interestingly, the other two cases which the authors did not classify as having a definite TBI history had a known history of epilepsy, which has been described to be associated with TBI sufficient to produce CTE pathology.17,18 In order to investigate sensitivity of the three-block CTE screening protocol, the authors also applied the protocol to materials from twelve additional cases of known CTE from the Australian Sports Brain Bank; they found that the protocol was able to detect CTE definitively in nine cases (75%) and in one other case the authors felt there were features that would have been suggestive enough to spur more comprehensive evaluation after initial screening. Ultimately, and while acknowledging that the three-block screening was imperfect and may have resulted in a small number of missed cases, the authors demonstrate further evidence that CTE is uncommon in the general community and particularly in the absence of known or implied TBI history. They also provide evidence against the notion that alcohol or substance abuse is a factor in the pathogenesis of CTE pathology. Chronic traumatic encephalopathy is not the “invisible wound” of war Addressing CTE in another critical community, in 2022 we reported the first large-scale analysis for CTE in the brains of military personnel.19 Our laboratory is the Department of Defense/Uniformed Services University Brain Tissue Repository, which is a brain bank exclusively dedicated to the collection and study of brain specimens derived from Service Members. We accept and comprehensively evaluate brain donations from anyone who has served in the military, irrespective of TBI history, neuropsychiatric disease, or other factors. Among our primary missions is identifying the underlying pathology for the so-called “invisible wound” of war: the frequent development of persistent cognitive, behavioral, and/or motor symptomatology following exposure to combat-related, and particularly blast-related, TBI which by routine head imaging shows no consistent anatomic defects. For our report, published in the New England Journal of Medicine, we evaluated 225 consecutive military brain donations involving a cohort with an age range of 18-87 years (average age of death: 48 years) and representing active duty and retired military personnel from all service branches. Represented were high frequencies of known military TBI exposure (20% with blast exposure, 9.3% with military impact-type TBI unassociated with blast), contact sports history (26.7%), and history of significant impact-type TBIs in civilian life unrelated to sports play (e.g. from motor vehicle accidents, assaults, falls leading to concussion, loss of consciousness/coma, skull injury, intracranial bleeding, and/or significant post-traumatic sequelae). Further, the cohort had high rates of psychiatric disease (39.1%, most commonly post-traumatic stress disorder), alcohol and/or substance abuse (43.1%), and death by suicide (22.7%); these rates are unfortunately comparable to reported rates in active duty and/or retired military populations.20-24 We blindly assessed all cerebral-cortex containing, AT8-immunostained slides that were available for each case, which amounted to a degree of evaluation that met or far exceeded current diagnostic guidelines for CTE10 (average: sections from 13 blocks per case, range: 5-23). We identified CTE pathology in ten of the 225 cases (4.4%). As can be inferred, CTE pathology did not correspond to the large majority of cases involving psychiatric disease, alcohol and/or substance abuse, and/or suicide. Further, when CTE was identified, it tended to be mild, or only barely diagnostic: five of the ten CTE cases only involved a single CTE pathognomonic lesion. Importantly, all ten of the CTE cases identified in our series involved a past history of participation in contact sports with or without an additional history of significant civilian impact-type TBI unrelated to sports (e.g. motor vehicle accident, physical assault, etc.). The relative risks for civilian TBI exposures, especially contact sports participation (relative risk not computable owing to 100% frequency of contact sport history in CTE cases; 95% CI: 6.16 to infinity) were substantially higher than those for military exposures, especially blast exposure. Though our study was underpowered precisely because there were few cases of CTE, in comparison to impact-type TBI exposures evaluated in our study, blast exposure history was the only TBI measure that had no significant association with CTE pathology (relative risk: 1.71; 95% CI: 0.46-6.37). Ultimately, our study suggests that CTE is relatively uncommon in the brains of military personnel, and when identified is overwhelmingly in the context of civilian TBI exposures, namely contact sports participation, and therefore is potentially unrelated to a military occupation at large. Further, CTE does not seem to account for the high rates of alcohol and/or substance abuse, psychiatric diagnoses suffered by Service Members. Finally, our series also suggests that blast exposure is not an independent risk factor for CTE development, though further research is needed. Relevance of APOE Gene Status and Chronic Traumatic Encephalopathy? Because not all of those who are exposed to impact-type TBI develop CTE pathology, even when repetitive and even in the context of contact sports participation, it certainly stands to reason that other risk factors or predispositions may be involved in pathogenesis. To date, pursuits toward the identification of potential genetic risk factors for CTE have been relatively lacking. However, in 2022 researchers Atherton et al. published findings on the “Association of APOE Genotypes and Chronic Traumatic Encephalopathy” in the journal JAMA Neurology.25 A total of 364 consecutive brain donations sourced from the Veterans Affairs-Boston University-Concussion Legacy Foundation Brain Bank were utilized in the study, including from 294 donors whose brains had known CTE pathology and 70 controls with histories of repetitive TBI exposure but no CTE pathology, all aged 47-77 years at death. The overall allele frequency for APOEε4 was 0.20. While the researchers found no significant association between CTE status and APOE genotype, they did find statistically significant associations between APOEε4 status and CTE stage (McKee stages I-IV) when considering donors aged >65 years, and even when adjusted for ADNC. The authors also suggest through from their statistical assessment that APOEε4 status is associated with a similar risk of a given CTE stage as that from playing more than 7 years of American football. Further, the researchers report an association between APOEε4 status and dementia in repetitive TBI-exposed individuals >65 years of age, however this association is lost after adjusting for ADNC. Regardless, the authors ultimately conclude that they have provided evidence that APOEε4 status may be a risk factor for CTE pathological and clinical outcomes, and imply that knowledge of APOEε4 status may be relevant towards precision medicine and with regard to individual decision-making on contact sport participation. Obviously, more research using larger sample sizes, prospectively gathered materials for this purpose, and performed in other independent laboratories will be necessary to validate and support these suggestions. There are a number of important concerns that may emerge from this impactful paper with regard to interpreting the findings. First and foremost, there were no statistically significant associations between APOEε4 status and outcomes of CTE status (i.e. yes or no), CTE stage, or clinical dementia in repetitive TBI-exposed individuals ≤65 years-old. While the authors excluded cases with ADNC to support their conclusions for individuals aged >65 years, this does not account for age-related ptau accumulation that may be incidental nor does it account for the 22 total cases with diagnoses of frontotemporal lobar dementia-tau that are reported to be within the study; certainly, these factors may impact CTE staging efforts. Further, in addition to analyzing associations with CTE stage, the researchers also performed a digital, quantitative assessment of ptau burden in dorsolateral prefrontal samples. While the authors calculated statistically significant p-values for the association of APOEε4 status and this quantitative measure of ptau burden, the relatively weak 1.04 OR after excluding ADNC cases is coupled with a 95% CI of 0.48-1.60. Interestingly, and unexpectedly, the results from weighing APOEε4 status with the quantitative assessment of ptau burden also did not correlate with the results analyzing CTE stage with respect to APOEε4 status; granted, while the assessment of ptau burden was performed only in the dorsolateral prefrontal cortex, this lack of correlation may nonetheless have implications regarding the McKee CTE staging scheme. Finally, what we found most interesting was the authors’ assessment of only former American football players within the study, which included a total of 323 cases. In this particular cohort, the authors found no significant association between duration of play and outcomes of clinical dementia or quantitative ptau burden in the dorsolateral prefrontal lobe. The authors did report statistical associations between duration of play and CTE status and stage, however these were surprisingly quite weak. The OR for CTE status and years of play was 1.25 (95% CI: 1.13-1.38) for individuals aged ≤65 and 1.32 (95% CI: 1.13-1.55) for individuals aged >65 after exclusion of ADNC cases. This data appears to suggest that our widely held concepts regarding the clinicopathologic correlation of this disease continue to need further investigation. Evidence of Sodium Channelopathy and Altered Node of Ranvier Structure following Concussive Traumatic Brain Injury Little is understood regarding the physiology of the immediate axonal response to TBI, including pathologic alterations such as the formation of axonal swellings and other processes that may be related to or co-occur with diffuse axonal injury (DAI). Further, a comprehensive understanding of pathophysiologic mechanisms underlying concussion and post-concussive symptoms and recovery continues to elude us. In 2022, authors Song et al. reported their use of a miniature swine model, relevant due to their gyrencephalic brain structure, for concussion/DAI to study alterations in axonal voltage-gated sodium channels and the structure of the nodes of Ranvier (NORs) after acute TBI.26 Nine animals were subjected to rotational TBI and sacrificed at different time intervals following injury (6 hours, 72 hours, and 2 weeks) and four served as controls. In parallel, researchers also studied human brain tissues derived from five individuals who died after acute, moderate-to-severe TBI in time intervals that were ≤14 days after injury (survival time ranged from 23 hours to 14 days, median survival time was 3 days), along with five sex- and age-matched controls. In both the pig and human materials, the authors confirmed evidence of DAI via demonstration of increased in APP immunolabeling in all injured tissues, and not in control tissues. From there, the authors used immunofluorescent antibodies targeting sodium channels within the NORs, using antibody to NaCh Isoform 1.6 (Nav1.6), as well as antibodies targeting other axonal proteins including Caspr, which is an axonal membrane protein accentuated in the paranodal region, and axonal cytoskeletal proteins βIV-spectrin, AnkG, and Nfasc186, which are more highly expressed within the NOR itself. In the pigs, the authors demonstrated marked and statistically significant loss of Nav1.6 labeling, indicating axonal sodium channel loss, at 6- and 72-hours following injury, with a recovery to the level of control animals at 2 weeks following injury. In association with sodium channel loss, the authors noted significant increase in NOR length 72-hours following injury, with partial recovery at 2 weeks, along with formation of so-called “void nodes” (defined by the complete loss of Nav1.6 with normal paranodal Caspr labeling) and “heminodes” (defined by loss of Caspr on a single side of the NOR, associated with normal or reduced Nav1.6). Finally, the authors noted loses of βIV-spectrin, ankyrin G, and neurofascin 186 within the NORs following acute TBI, with evidence diffusion of the proteins into the paranodal space. The above observations were also noted in the human tissues, which demonstrated marked loses of Nav1.6 and similar changes to the structure of the NORs and analyzed proteins. Considering the vital role of voltage-gated sodium channels and NOR structure for the generation of action potentials, these findings provide critical evidence that neurologic dysfunction following concussion, or other TBI, may in part be due to sodium channelopathy and altered nerve conduction by these mechanisms. Further, researchers in the study also found that many of the NOR changes were observed adjacent to APP-positive axonal swellings, including at the 6-hour mark of survival in the swine model. This observation potentially indicates new and potentially more sensitive biomarkers by which we can detect DAI. The role of TDP-43 in Exacerbating Neuropathology and Outcomes Following Traumatic Brain Injury Beyond tau there is increasing interest in the role of other known neurodegenerative proteins, whether directly or otherwise, involved in neuropathologic and ultimately clinical outcomes following TBI(s). Moreover, mechanisms by which TBI may be related to exacerbated neurodegenerative pathologies and/or multiple neurodegenerative pathologies are a highly active area of investigation. In 2022, authors Gao et al. reported their findings from a study focused on the influence of TDP-43 in driving ADNC and cognitive impairment in mice following TBI.27 As part of their impressive study, the authors utilized 5xFAD APP transgenic mice exposed to a model of mild closed head injury using a well-published, commercially available impact device. Trauma was induced at 2 months of age. Via novel object recognition assessment and a maze test, the researchers found that a single mild TBI exposure was associated with the accelerated development of memory and cognitive deficits in the APP transgenic mice, compared to non-exposed APP transgenic mice and APP wildtype mice; the deficits in the APP transgenic mice were recognized at 3 months of age, whereas typically these mice do not show such deficits until beyond 5 months of age. Further, the authors demonstrate significantly increased deposits of Aβ plaques and significantly increased expressions of ptau, acetylated tau, β-site amyloid precursor protein cleaving enzyme 1 (BACE1, an enzyme implicated in Aβ plaque formation), and TDP-43 in comparison to non-exposed APP transgenic mice and APP wildtype mice. It is interesting to note that APP transgenic mice, as they age, do not develop tauopathy. Further, via reverse transcription and real-time polymerase chain reaction analysis performed on harvested hippocampal tissue 24 hours after TBI exposure, the authors report significantly elevated inflammatory markers and pro-inflammatory cytokines (vimentin, TNFα, IL-1β, and IL-6) in APP transgenic mice following TBI. As such, the authors demonstrate that a single impact-type TBI exacerbates neuropathological and clinical deficits in mice predisposed to Aβ pathology, and notably among the neuropathological observations were increases in TDP-43, tau, and indices of neuroinflammation. The researchers then sought to address the question of how TDP-43 may be involved in the exacerbation of the neuropathological and clinical deficits noted in the APP transgenic mice. This was accomplished via a series of experiments inducing single and/or multiple TBIs following the injection of RNA products via viral vectors, which included those designed to silence endogenous TDP-43 expression, into the hippocampus of the APP transgenic mice. The authors report that cognitive and memory deficits, as well as neurodegenerative indices (including immunohistochemical assessment of Aβ plaques and western blot analyses including for Aβ, BACE1, ptau, and acetylated tau) and neuroinflammatory indices (immunohistochemical analysis for astrocytes [GFAP] and microglia [Iba1]) after single and repeated TBI in the APP transgenic mice were all prevented/alleviated following induction of TDP-43 knockdown in comparison to the control group, and were also reverted in a group which was administered a TDP-43 knockdown rescue protocol. Furthermore, APP transgenic mice injected with a viral vector to induce TDP-43 overexpression alone, without TBI, also demonstrated accelerations in memory and cognitive impairments, mimicking those who had sustained TBI. These cases also showed dramatic increases in indices including BACE1, acetylated tau, ptau, phosphorylated glycogen synthase kinase-3β (pGSK3β, an enzyme known to play an important role in tau phosphorylation and ADNC) and phosphorylated NF-κB, and also increased labeling of astrocytes and microglia. Other components of the study demonstrate evidence that TDP-43 overexpression in APP transgenic mice, whether induced by TBI or by viral vector injection, downregulates the expression of synaptic proteins including various glutamate receptor subunits and PSD95, and thus interferes with synaptic integrity. Finally, the researchers report findings investigating the role of TDP-43 in tau phosphorylation and Aβ deposition through work with cultured hippocampal neurons from tau transgenic mice and APP transgenic mice. They report evidence that TDP-43 knockdown in tau transgenic neuronal cells is associated with reduced expression of ptau and pGSK3β, and also that TDP-43 is upstream from GSK3β in driving tau phosphorylation. In neuronal cells from APP transgenic mice, the researchers report that TDP-43 knockdown was associated with reduced expression of BACE1 which provides further evidence that TDP-43 has a driving role in Aβ formation. Ultimately, the extensive and thorough research highlighted by this study provides fascinating evidence that not only does TDP-43 play an instrumental role in driving both tau phosphorylation and Aβ formation in the context of AD, but also may be a principle link that connects TBI with the subsequent development of neurodegenerative disease. In Memoriam: John Q. Trojanowski, M.D., Ph.D. We conclude this year’s listing on a solemn note, but also in awe of the legacy left by a titan in the field of neurodegeneration, neuropathology in general, and in the context of this review, neurotrauma in particular. On February 8th, 2022, we lost Dr. John Q. Trojanowski due to fatal complications from a fall. Dr. Trojanowski received his M.D. and Ph.D. degrees from Tufts University in 1976. Following this, he completed residency training in pathology and fellowship training in neuropathology at the Massachusetts General Hospital and the University of Pennsylvania, respectively. He stayed in Pennsylvania and followed his training with an extraordinary career that included the establishment and direction of the University of Pennsylvania Center of Neurodegenerative Disease Research Center with his wife and scientific partner, Dr. Virginia Lee (Figure 3), co-authorship in over 1,400 peer-reviewed papers, an abundance of accolades, and an immeasurable degree of influence that he impressed upon a lifetime’s worth of trainees, other mentees, and professional colleagues. Pertaining to work in neurotrauma, John was one of the foundational members of the Collaborative Neuropathology Network Characterizing ouTcomes of TBI (CONNECT-TBI).28 He was a co-author on several important and highly cited studies concerning pathologic protein accumulation due to TBI in both animal and human studies from the late 1990s and early 2000s.29-34 More recently he had been involved in CTE research which included but certainly was not limited to a major 2020 study published in Brain which established that the immunophenotypes of astroglial and neuronal ptau in cases of CTE are distinct and individually resemble age-related tau astrogliopathy (4R-tau immunoreactive, thorn-shaped astrocytes) and ADNC (3R and 4R tau), respectively.35 John’s life and legacy are beautifully memorialized in writings by several of our peers and in the media36-42, and we strongly encourage readers to visit these remembrances. >
Figure 3. Dr. John Q. Trojanowski (right) and his wife and scientific partner, Dr. Virginia M. Lee (left). Together, John and Virginia Co-founded and directed the Center for Neurodegenerative Disease Research at the University of Pennsylvania Acknowledgements We would like to thank Ms. Sofia Echelmeyer in the office of External Affairs and University Media Services at the Uniformed Services University for her magnificent illustrative work (Figure 1). We also thank Dr. Edward B. Lee from the Department of Pathology and Laboratory Medicine at the University of Pennsylvania for providing a beautiful photograph of Drs. John Trojanowski and Virginia Lee (Figure 3). Disclaimer The information/content, conclusions, and/or opinions expressed herein do not necessarily represent the official position or policy of, nor should any official endorsement be inferred on the part of, Uniformed Services University, the Department of Defense, the US Veterans Administration, the U.S. Government or the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc. Conflicts of Interest The authors do not have any conflict of interest to declare. References 1. Hill AB. The Environment and Disease: Association or Causation? Proc R Soc Med. 1965 May;58(5):295-300. PMCID: PMC1898525 2. Nowinski CJ, Bureau SC, Buckland ME, et al. Applying the Bradford Hill Criteria for Causation to Repetitive Head Impacts and Chronic Traumatic Encephalopathy. Front Neurol. 2022 Jul 22;13:938163. PMCID: PMC9355594 DOI: 10.3389/fneur.2022.938163 3. Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol 2012;22:142–9. PMCID: PMC3979351 DOI: 10.1111/j.1750-3639.2011.00513.x 4. Shively SB, Edgerton SL, Iacono D, et al. Localized cortical chronic traumatic encephalopathy pathology after single, severe axonal injury in human brain. Acta Neuropathol 2017;133(3):353-366. PMCID: PMC5325841 DOI: 10.1007/s00401-016-1649-7 5. Iverson GL, Gardner AJ, Shultz SR, et al. Chronic traumatic encephalopathy neuropathology might not be inexorably progressive or unique to repetitive neurotrauma. Brain. 2019;142(12):3672-3693. PMCID: PMC6906593 DOI: 10.1093/brain/awz286 6. Ackermans NL, Varghese M, Williams TM, et al. Evidence of traumatic brain injury in headbutting bovids. Acta Neuropathol. 2022;144(1):5-26. PMCID: PMC9217783 DOI: 10.1007/s00401-022-02427-2 7. Kerwin J, Yücesoy A, Vidhate S, et al. Sulcal Cavitation in Linear Head Acceleration: Possible Correlation With Chronic Traumatic Encephalopathy. Front Neurol. 2022 Feb 28;13:832370. PMCID: PMC8918564 DOI: 10.3389/fneur.2022.832370 8. Miller ST, Cooper CF, Elsbernd P, Kerwin J, Mejia-Alvarez R, Willis AM. Localizing Clinical Patterns of Blast Traumatic Brain Injury Through Computational Modeling and Simulation. Front Neurol. 2021 May 20;12:547655. PMCID: PMC8173077 DOI: 10.3389/fneur.2021.547655 9. Shively SB, Horkayne-Szakaly I, Jones RV, Kelly JP, Armstrong RC, Perl DP. Characterisation of interface astroglial scarring in the human brain after blast exposure: a post-mortem case series. Lancet Neurol. 2016 Aug;15(9):944-953. DOI: 10.1016/S1474-4422(16)30057-6 10. Bieniek KF, Cairns NJ, Crary JF, et al. The Second NINDS/NIBIB Consensus Meeting to Define Neuropathological Criteria for the Diagnosis of Chronic Traumatic Encephalopathy. J Neuropathol Exp Neurol. 2021;80(3):210-219. PMCID: PMC7899277 DOI: 10.1093/jnen/nlab001 11. Butler MLMD, Dixon E, Stein TD, et al. Tau Pathology in Chronic Traumatic Encephalopathy is Primarily Neuronal J Neuropathol Exp Neurol. 2022;81(10):773-780. PMCID: PMC9487650 DOI: 10.1093/jnen/nlac065 [published correction appears in J Neuropathol Exp Neurol. 2023 Mar 20;82(4):373. PMCID: PMC10025875 DOI: 10.1093/jnen/nlac125] 12. Ameen-Ali KE, Bretzin A, Lee EB, et al. Detection of astrocytic tau pathology facilitates recognition of chronic traumatic encephalopathy neuropathologic change. Acta Neuropathol Commun. 2022;10(1):50. PMCID: PMC8996534 DOI: 10.1186/s40478-022-01353-4 13. Bieniek KF, Ross OA, Cormier KA, et al. Chronic traumatic encephalopathy pathology in a neurodegenerative disorders brain bank. Acta Neuropathol 2015;130(6):877-89. PMCID: PMC4655127 DOI: 10.1007/s00401-015-1502-4 14. Forrest SL, Kril JJ, Wagner S, et al. Chronic Traumatic Encephalopathy (CTE) Is Absent From a European Community-Based Aging Cohort While Cortical Aging-Related Tau Astrogliopathy (ARTAG) Is Highly Prevalent. J Neuropathol Exp Neurol 2019;78(5):398-405. DOI: 10.1093/jnen/nlz017 15. Postupna N, Rose SE, Gibbons LE, et al. The Delayed Neuropathological Consequences of Traumatic Brain Injury in a Community-Based Sample. Front Neurol 2021;12:624696. PMCID: PMC8008107 DOI: 10.3389/fneur.2021.624696 16. Suter CM, Affleck AJ, Lee M, et al. Chronic Traumatic Encephalopathy in a Routine Neuropathology Service in Australia. J Neuropathol Exp Neurol. 2022;81(10):790-795. DOI: 10.1093/jnen/nlac071 17. Geddes JF, Vowles GH, Nicoll JA, et al. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol 1999;98:171-8. DOI: 10.1007/s004010051066 18. Thom M, Liu JY, Thompson P, et al. Neurofibrillary tangle pathology and Braak staging in chronic epilepsy in relation to traumatic brain injury and hippocampal sclerosis: a post-mortem study. Brain. 2011;134(Pt 10):2969-2981. PMCID: PMC3187539 DOI: 10.1093/brain/awr209 19. Priemer DS, Iacono D, Rhodes CH, Olsen CH, Perl DP. Chronic Traumatic Encephalopathy in the Brains of Military Personnel. N Engl J Med. 2022;386(23):2169-2177. DOI: 10.1056/NEJMoa2203199 20. Howard JT, Stewart IJ, Amuan M, Janak JC, Pugh MJ. Association of traumatic brain injury with mortality among military veterans serving after September 11, 2001. JAMA Netw Open 2022;1;5(2): e2148150. PMCID: PMC8837911 DOI: 10.1001/jamanetworkopen.2021.48150 21. Richardson LK, Frueh BC, Acierno R. Prevalence estimates of combat-related post-traumatic stress disorder: critical review. Aust N Z J Psychiatry 2010;44(1):4-19. PMCID: PMC2891773 DOI: 10.3109/00048670903393597 22. Tanielian T, Jaycox L. (Eds.). Invisible Wounds of War: psychological and cognitive injuries, their consequences, and services to assist recovery. Santa Monica, CA: RAND Corporation, 2008. (https://www.rand.org/pubs/monographs/MG720.html). 23. Meadows SO, Engel CC, Collins RL, et al. 2015 Health Related Behaviors Survey: substance use among U.S. active-duty Service Members. Santa Monica, CA: RAND Corporation, 2018. (https://www.rand.org/pubs/research_briefs/RB9955z7.html) 24. Teeters JB, Lancaster CL, Brown DG, Back SE. Substance use disorders in military veterans: prevalence and treatment challenges. Subst Abuse Rehabil 2017;8:69-77. PMCID: PMC5587184 DOI: 10.2147/SAR.S116720 25. Atherton K, Han X, Chung J, et al. Association of APOE Genotypes and Chronic Traumatic Encephalopathy. JAMA Neurol. 2022;79(8):787-796. PMCID: PMC9237800 DOI: 10.1001/jamaneurol.2022.1634 [published correction appears in JAMA Neurol. 2023 Feb 1;80(2):215. PMCID: PMC9577875 DOI: 10.1001/jamaneurol.2022.3673] 26. Song H, McEwan PP, Ameen-Ali KE, et al. Concussion leads to widespread axonal sodium channel loss and disruption of the node of Ranvier. Acta Neuropathol. 2022;144(5):967-985. DOI: 10.1007/s00401-022-02498-1 27. Gao F, Hu M, Zhang J, Hashem J, and Chen C. TDP-43 drives synaptic and cognitive deterioration following traumatic brain injury. Acta Neuropathol. 2022 Aug;144(2):187-210. PMCID: PMC9945325 DOI: 10.1007/s00401-022-02449-w 28. Smith DH, Dolle JP, Ameen-Ali KE, et al. the Collaborative Neuropathology Network Characterizing ouTcomes of TBI (CONNECT-TBI). Acta Neuropathol Commun. 2021;9(1):32. PMCID: PMC7919306 DOI: 10.1186/s40478-021-01122-9 29. Smith DH, Uryu K, Saatman KE, Trojanowski JQ, McIntosh TK. Protein accumulation in traumatic brain injury. Neuromolecular Med. 2003;4(1-2):59-72. DOI: 10.1385/NMM:4:1-2:59 30. Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol. 2009;19(2):214-223. PMCID: PMC3014260 DOI: 10.1111/j.1750-3639.2008.00176.x 31. Ikonomovic MD, Uryu K, Abrahamson EE, et al. Alzheimer's pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol. 2004;190(1):192-203. DOI: 10.1016/j.expneurol.2004.06.011 32. Uryu K, Laurer H, McIntosh T, et al. Repetitive mild brain trauma accelerates Abeta deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. J Neurosci. 2002;22(2):446-454. PMCID: PMC6758680 DOI: 10.1523/JNEUROSCI.22-02-00446.2002 33. Uryu K, Chen XH, Martinez D, et al. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol. 2007;208(2):185-192. PMCID: PMC3979356 DOI: 10.1016/j.expneurol.2007.06.018 34. Smith DH, Chen XH, Nonaka M, et al. Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999;58(9):982-992. DOI: 10.1097/00005072-199909000-00008 35. Arena JD, Smith DH, Lee EB, Gibbons GS, Irwin DJ, Robinson JL, Lee VM, Trojanowski JQ, Stewart W, Johnson VE. Tau immunophenotypes in chronic traumatic encephalopathy recapitulate those of ageing and Alzheimer’s disease. Brain. 2020 May 1;143(5):1572-87. PMCID: PMC7241956 DOI: 10.1093/brain/awaa071 36. Lee EB. John Q. Trojanowski: neuropathology icon. Acta Neuropathol. 2022 Apr;143(4):419-425. PMCID: PMC10259176 DOI: 10.1007/s00401-022-02413-8 37. Lee EB. The continuing legacy of John. Acta Neuropathol. 2022;144(6):1063-1064. DOI: 10.1007/s00401-022-02514-4 38. Nelson PT, Lee EB, Judkins AR. John Q. Trojanowski, MD, PhDDecember 17, 1946–February 8, 2022. J Neuropathol Exp Neurol. 2022 Jun 1;81(6):434-6. DOI: 10.1093/jnen/nlac029 39. Chen-Plotkin AS. John Q. Trojanowski, 'tour de force' in neurodegeneration (1946-2022). Nat Neurosci. 2022;25(6):675-676. DOI: 10.1038/s41593-022-01087-5 40. Irwin DJ, Lee EB. A tribute to John Q. Trojanowski (1946-2022). J Clin Invest. 2022 May 16;132(10):e161019. PMCID: PMC9106356 DOI: 10.1172/JCI161019 41. Lashuel HA. Remembering John Q Trojanowski, in his own words: A life dedicated to discovering building blocks and using them to build bridges of knowledge, collaboration, and discovery. NPJ Parkinsons Dis. 2022;8(1):43. PMCID: PMC9005626 DOI: 10.1038/s41531-022-00310-1 42. Kolata G. John Q. Trojanowski Dies at 75; Changed Understanding of Brain Diseases. The New York Times, 2022 Mar 1 (https://www.nytimes.com/2022/03/01/health/john-q-trojanowski-dead.html).
Copyright: © 2023 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |