|
|
|
Free Neuropathology 3:12 (2022) |
|
Review |
|
Neuropathology and epilepsy surgery: 2022 update |
|
Ingmar Blümcke1 |
|
1 Department of Neuropathology, University Hospital Erlangen, Germany |
|
Corresponding author: |
|
Submitted: 17 March 2022 Accepted: 25 April 2022 Copyedited by: Vanessa Goodwill Published: 03 May 2022 |
|
Keywords: Brain, Seizure, Malformation, Brain tumor, Molecular diagnostics |
|
Abstract The impact of a precise histopathology diagnosis and molecular workup for surgical patient management remains a controversial issue in epileptology with a lack of diagnostic agreement as root cause. Very recent advances in genotype-phenotype characterization of epilepsy-associated developmental brain lesions, including the first diagnostically useful DNA methylation studies, opened new avenues and will help to finally resolve these issues. A series of most recent articles were decisively selected by the author to exemplify the areas of improvement in neuropathology and epilepsy surgery. These topics include the progress in genotype-phenotype association studies of Focal Cortical Dysplasia (FCD) leading to the discovery of new molecularly defined entities, i.e. mild malformation of cortical development with oligodendroglial hyperplasia in epilepsy (MOGHE), SLC35A2 altered. These studies also triggered the first update of the international FCD consensus classification scheme from 2011, which will hopefully support diagnostic agreement in clinical practice and research. The dilemma of new tumor entities proposed by the 5th edition of the WHO classification primarily associated with early seizure onset yet not well introduced to the epileptology community will also be discussed in the light of emerging experimental evidence when transfecting the developing murine brain with the single most important genetic alteration for both carcino- and epileptogenesis, i.e. BRAF V600E. Introduction The impact of a precise histopathology diagnosis and molecular workup for patient management remains a controversial issue in epileptology 1, i.e., the decision making for surgical treatment when a patient with focal epilepsy is considered drug-resistant and prediction of postsurgical seizure freedom should be based on the underlying cause. This discussion continues despite large enough patient series suggesting a positive prediction of scientifically defined brain lesions for long-term postsurgical seizure control for more than 5 years2. A root cause of these inconsistencies is likely due to lack of diagnostic agreement across histopathology laboratories around the world, a fact widely recognized in the scientific literature3-7. The introduction of molecular diagnostics has helped to overcome this issue in the field of neurooncology with the best renowned examples of abandoning mixed oligo-astrocytomas and introducing molecularly defined and clinically/therapeutically relevant tumor subtypes. Literature published in 2021 about the topic of neuropathology and epilepsy surgery elicited new hope for epilepsy centers around the world to better connect with molecular diagnostics and integrated genotype-phenotype pathology diagnosis, which will finally move the field toward personalized medicine and targeted treatment options. The following chapters will address ten topics of interest to the author based on papers published in 2020-2022 and with reference to other older topic-related key data. TOPIC 1: The molecular phenotype of human epilepsies at the single cell level TOPIC 2: Brain somatic mutations in cortical malformations (MCD) TOPIC 3: DNA-methylation classifier for malformations of cortical development TOPIC 4: New clinico-pathologically and genetically defined entities: MOGHE and FCD1A TOPIC 5: Toward a first update of the international consensus classification of FCD TOPIC 6: News from low-grade epilepsy-associated brain tumors (LEAT): human studies TOPIC 7: News from LEAT: animal models TOPIC 9. The fine structure of the epileptogenic neocortex and white matter TOPIC 10: What is still missing to further the advancement of neuropathology and epilepsy surgery?
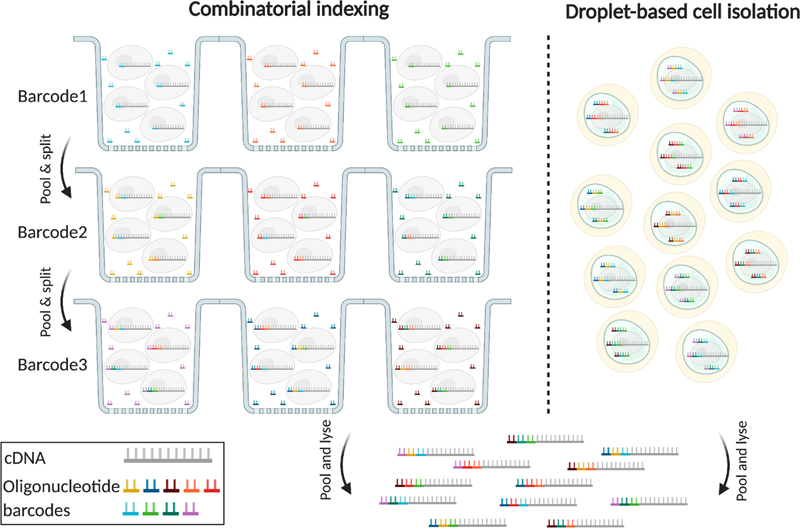
TOPIC 1: The molecular phenotype of human epilepsies at the single cell level The inception of single cell biology has completely revolutionized the study of neurological diseases and has the potential to answer many of the yet unresolved questions in the neurosciences8. The advancement in molecular-genetic barcoding technology (Figure 1) at the single cell level including single-cell genomics and transcriptomics finally reached the realm of epileptology. Human surgical tissue samples of clinically well-characterized patients have ever been a valuable resource for research, although vast differences in the patients’ genetic background, clinical histories and medical treatment pose major challenges to comprehensive data analysis. Focusing on single cell genomics, transcriptomics, and finally also epigenomics in focal epilepsy research, will circumvent the averaging artifact when studying bulk brain tissue and will reveal the kind of granularity that is needed for investigating the consequences of network alterations at the single cell level8.
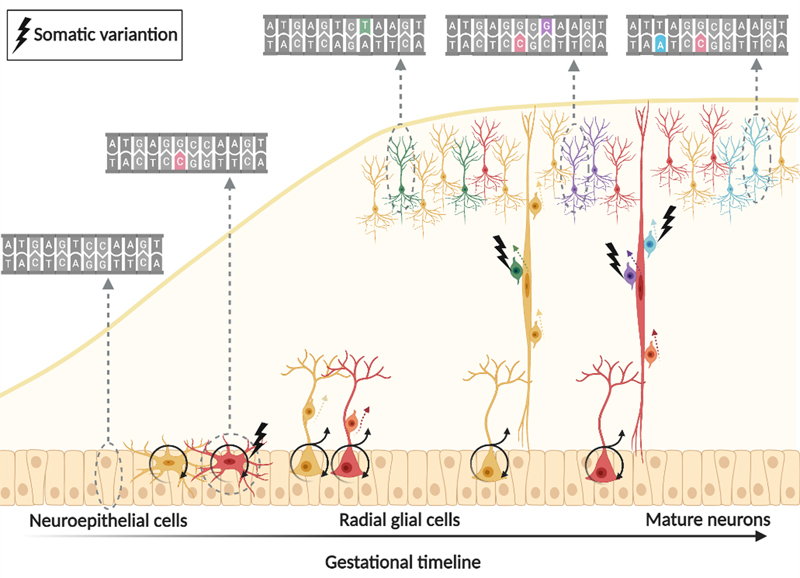
Figure 1: Single-cell genomic applications using single cell combinatorial indexing or droplet based cell isolation with barcoding 8 Single cell combinatorial indexing (SCI) and droplet-based cell isolation are the most popular barcoding strategies used by most high-throughput single cell genomic applications. SCI uniquely tags the nucleic acid molecules in each cell through serial mixing, splitting, and barcoding. The higher the number of barcoding steps, the higher the number of cells that can be uniquely tagged in each experiment. Droplet-based techniques rely on physical isolation of individual cells and engineered barcoded beads in nanoliter droplets, which limits their scalability but they produce less noisy results. With permission from the author 8. Whereas single cell genomics have already been applied using human epilepsy surgery brain samples9, Pfisterer and colleagues described in 2020 for the first time single nucleus transcriptomics in human temporal lobe epilepsy and non-epileptic subjects10. They deciphered dysfunctional neuronal subtypes in the seizure active cortical region and found that the largest transcriptomic changes occurred in distinct subtypes of principal neurons, as classified by the layer-specific marker genes CUX2, RORB, THEMIS, and FEZF2, and GABAergic interneurons, as classified by the marker genes PVALB, SST, VIP, and ID2. As a result, FEZF2 positive neurons of layers 5 and 6 and CUX2 neurons of layers 2 and 3 were more affected than other subtypes of the same families. Similarly, parvalbumin and somatostatin interneurons showed more of an epileptogenic signature than other GABAergic interneurons. Furthermore, the subtypes with the largest epilepsy-related transcriptomic changes belong to same circuits. In addition, glutamate signaling exhibited a strong dysregulation in epilepsy, as indexed by layer-specific transcriptional changes in multiple glutamate receptor genes and upregulation of genes coding for AMPA receptor auxiliary subunits10. Overall, these data confirmed neuron-specific molecular phenotypes in the epileptic neocortex, which may become novel targets for anti-epileptic precision medicine in the near future. Over the past 5 years DNA-methylation has gained significant interest and momentum in neuropathology. The development of DNA-methylation based classifier for epilepsy-associated brain malformations will be discussed further below (see TOPIC 3 for further reading). DNA methylation has not yet been addressed at the single cell level in human neurosurgical specimens though. In the mouse brain, however, a first comprehensive assessment of the epigenomes of mouse brain cell types was published in 202111. The researchers used single-nucleus DNA methylation sequencing from 45 regions of the mouse cortex, hippocampus, striatum, pallidum and olfactory areas and identified 161 cell clusters with distinct spatial locations and projection targets11. Furthermore, they constructed taxonomies of these epigenetic types, annotated with signature genes, regulatory elements and transcription factors. These features revealed the repetitive usage of regulators in excitatory and inhibitory cells to determine cellular subtypes. Furthermore, an artificial neural network model was developed to predict neuron cell-type identity and their spatial brain localization. The creation of a comprehensive DNA methylation-based atlas will establish the epigenetic basis for neuronal diversity and spatial organization throughout the mammalian brain, being also instrumental when developing a human methylome brain atlas in the near future, including epilepsy surgery brain samples from various disorders, e.g. cortical malformations and hippocampal sclerosis. TOPIC 2: Brain somatic mutations in cortical malformations Fifty years ago, in 1971, Taylor and colleagues coined the term Focal Cortical Dysplasia (FCD) for peculiar lesions causing drug resistant epilepsy12, which is now the most common cause for epilepsy surgery in children13. The study of resected human FCD tissue using advanced genomic technologies (Figure 1) has led to remarkable advances in understanding the genetic basis of FCD (Figure 2)14. Mechanistic parallels have emerged between these malformative lesions and low-grade and epilepsy-associated developmental brain tumors (LEAT). Two comprehensive review articles published in 2021 and 2022 recapitulated the avenue from recognition of somatic variants in genes related to cell growth, i.e. the mTOR pathway in FCD Type 2, which were acquired in neuronal progenitors during neurodevelopment14,15. The timing of the genetic event and the specific gene involved during neurodevelopment is likely to drive the nature and size of the lesion, and is maybe also related to the lobar localization. The proposed two-hit mechanism in patients presenting primarily with germline mutations, however, e.g., DEPDC5, has been scientifically approved but remains often difficult to detect with standard technologies. Along these lines, Lee and coworkers newly reported pathogenic brain-specific somatic Ras homolog enriched in brain (RHEB) variants in three patients with cortical malformations16. Interestingly, the somatic variant load directly correlated with the size of the malformation and upregulated mTOR activity was confirmed in dysplastic tissues. Laser capture microdissection showed enrichment of RHEB variants in dysmorphic neurons and balloon cells, i.e. FCD Type 2B. These findings added RHEB as an additional mTOR pathway gene to the diagnostically relevant gene panel for MCD. That the extent of dysplastic brain directly correlated with the somatic variant load suggested that cortical malformations with cytopathological features, i.e. FCD Type 2, represent a disease continuum from a regionally localized dysplasia to lobar or full blown hemimegalencephaly.
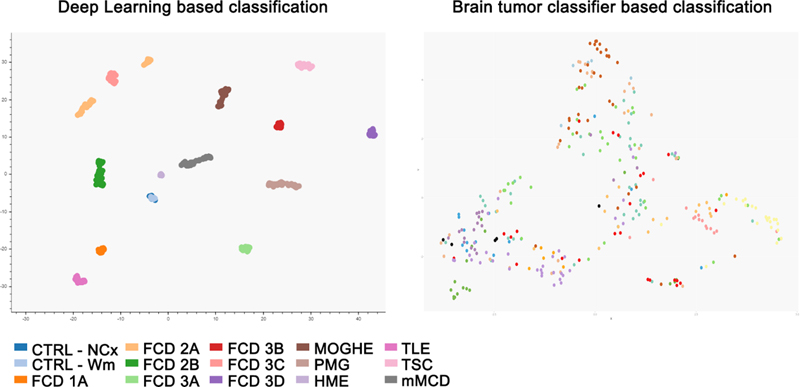
Figure 2: “One brain – many genomes” 17 Somatic variants are spontaneously acquired during neurodevelopment. All the somatic variants in a progenitor cell are passed down to its daughter cells. The number of cells carrying a specific variant is an indirect marker for the developmental time point at which it was generated. With permission from the author 8. Brain mosaicism, however, can only be detected in tissue obtained from autopsy or brain biopsy. Liquid biopsy using cell-free DNA derived from cerebrospinal fluid (CSF) could yet be another source for genetic testing, as successfully proven in malignant brain tumors. In 2021, Ye and coworker published a proof of principle study demonstrating that CSF liquid biopsy is valuable in investigating mosaic neurological disorders where brain tissue is unavailable18. First, they sequenced CSF cell-free DNA (cfDNA) in 28 patients with focal epilepsy and 28 controls (using droplet digital polymerase chain reaction (PCR)). They detected somatic mutations in three patients, i.e., the LIS1 p.Lys64* variant at 9.4% frequency in one patient with subcortical band heterotopia; the TSC1 p.Phe581His*6 variant at 7.8% frequency in another patient with FCD and thirdly the BRAF p.Val600Glu variant at 3.2% frequency in a patient with ganglioglioma. They also determined that cfDNA was brain-derived by using whole-genome bisulfite sequencing and demonstrating an enrichment of brain-specific DNA methylation patterns. In a second parallel study and publication, same results were obtained from CSF collected during epilepsy surgery19. Somatic variants were detected in cfDNA from 3 epileptic patients with known somatic mutations previously identified in brain tissue (out of 12 patients included in the study). Both proof-of-principle studies provide evidence that brain mosaicism can be detected in the CSF-derived cfDNA and open future avenues for detecting the mutant allele driving epilepsy in CSF. TOPIC 3: DNA-methylation classifier for malformations of cortical development The histopathological work-up of epilepsy surgery specimens is often based only on routine hematoxylin and eosin (H&E) staining with its many difficulties and pitfalls in reliable identification of anatomical landmarks and cortical layering. This applies particularly to samples not resected anatomically en bloc, or when compromised by peri-operative artefacts or diagnostic procedures such as intracranial electroencephalography (EEG) recordings, laser ablation or thermo-coagulation. It has been a long-standing effort in histopathology, therefore, to support the microscopic diagnoses with objectifiable diagnostic measures. Indeed, the same issue applied to the scenario in neuro-oncology before the discovery and introduction of predictive and prognostic genetic markers for the differential diagnosis of high-grade glioma and embryonal brain tumors, which helped to develop a reliable and widely used open-access DNA methylation-based brain tumor classifier20. Jabari and coworkers presented such a first DNA methylation classifier for cortical malformations in 2021, which opens the avenue for an objective genotype-phenotype approach in epilepsy surgery using routinely processed (archival) formalin-fixed and paraffin embedded (FFPE) tissue samples21. This work included a series of 308 histopathologically defined samples to cover the broad spectrum of MCD, including most common FCD subtypes, as well as polymicrogyria, hemimegalencephaly, tuberous sclerosis complex, mild malformations of cortical development (mMCD), and MOGHE specimens as well as non-MCD epilepsy samples and non-epilepsy post-mortem controls. The possibility to apply such a DNA methylation classification for non-neoplastic lesions has been questioned and is an ongoing challenge due to the often low content of abnormal cells and admixture with architecturally normal-appearing cortical areas. The possibility to microscopically dissect an area of the lesion with the most cell dense abnormality from FFPE tissue blocks was an important foundation, therefore, to the success of this project. The study did also follow a classifier approach different from the well-established Heidelberg classifier for brain tumors. In fact, their paper disclosed a graph in the supplemental material showing how all of their MCD samples would assemble with the Heidelberg classifier (Figure 3). This highlights the challenge but also the benefit of advanced bioinformatics for developing a DNA methylation classifier. It also highlights the need for a disease-specific pipeline addressing structural brain lesions in epilepsy and potential confounders. As an example, seizures themselves alter the DNA methylation map as demonstrated in a recent in vitro culture model “epilepsy in a dish”22. The variable seizure burden in a given patient, the affected brain region (i.e. grey and/or white matter), and the surgical tissue sample (e.g., representing the lesion or its periphery), present an ongoing issue when using the DNA methylation methodology for human brain specimens. Assessing as many potential confounders in the new pipeline published by Jabari et al. helped to separate the histopathologically labelled groups into well distinguishable entities e.g., taking into account the different referral centres, seizure onset, disease duration before surgery, cellular heterogeneity, batch effects, and sex. Finally, a test cohort of 43 independent surgical samples from different epilepsy centres was used to test the precision of the DNA methylation-based MCD classifier. Amongst this cohort were 19 samples previously reviewed by an international consensus expert panel (will be discussed further below). All samples from the test cohort were accurately assigned to their histopathologically agreed disease classes by the algorithm. This report provides a first DNA methylation-based MCD classification scheme suitable across major histopathological disease entities observed in epilepsy surgery. There is still a lot of work to do, however. The impact of increasingly recognized brain somatic mutations in FCD and other MCD entities (see chapters above), were not yet addressed in this study and need further attention.
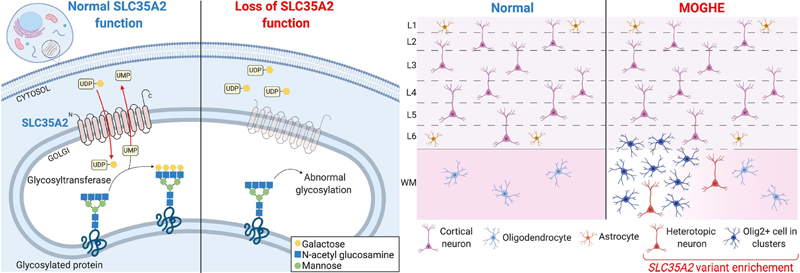
Figure 3: DNA methylation-based classifier using a new deep learning algorithm adapted for epilepsy surgery brain samples (on the left) compared to the Heidelberg brain tumor classifier (on the right) On the left: UMAP - Uniform Manifold Approximation and Projection of the decision boundaries of the trained deep learning algorithm showing the distribution of 308 datasets (taken from 21; this is an open access article distributed under the terms of the Creative Commons CC BY license). Colors correspond to the following coding CTRL – post-mortem control, NCx – neocortex, WM – white matter; FCD – Focal Cortical Dysplasia Type 1, 2 and 3 according to ILAE classification scheme; MOGHE - mild malformation with oligodendroglial hyperplasia in epilepsy, PMG - polymicrogyria, HME - hemimegalencephaly, TLE – temporal lobe epilepsy without cortical dysplasia, TSC - tuberous sclerosis complex, mMCD - mild malformations of cortical development. On the right: t-SNE – “t-distributed stochastic neighbour embedding” of the brain tumor classifier from Heidelberg of the same samples shown on the left. TOPIC 4: New clinico-pathologically and genetically defined entities: MOGHE and FCD1A In 2021, we witnessed the exciting rise of a new, genetically defined cortical malformation associated with early onset focal epilepsy. The term “mild malformation with oligodendroglial hyperplasia in epilepsy” (MOGHE) was already coined in 201723 but received only little attention by the histo-/neuropathology community. When epileptologists described its MRI fingerprint in 201924, many more reports became available cumulating in 2021 as a distinct clinico-pathological MOGHE pattern in children with early seizure onset and mostly affecting the frontal lobe25-28. Two other papers in 2021 almost simultaneously assigned brain somatic mutations in the UDP-galactose transporter SLC35A2 gene to MOGHE6,29. The SLC35A2 mutation was first discovered in a cohort of focal epileptic brain lesions in 2018, replicated in 2019 and originally assigned to either mMCD or FCD ILAE Type 1 entities30,31. Notwithstanding, both of these differential histopathology diagnoses are difficult and poorly defined. Bonduelle and coworker specifically searched for SLC35A2 mutations in histopathologically confirmed MOGHE and detected pathogenic mutations in 9 out of 20 new cases included in their series (45%). The frequency of somatic variants were usually above 10%, ranging from 1.4% to 52%. Droplet digital PCR of microdissected cells from one MOGHE case confirmed variant enrichment in abnormally clustering oligodendroglial cells and heterotopic neurons, suggesting that the mutation targeted neuroglial progenitors during brain development. These data provide compelling evidence for a consistent genotype-phenotype correlation in MOGHE with SLC35A2 mosaicism as a diagnostic and probably also prognostic marker, as more than 60% of MOGHE patients benefit from gross total resection of the lesion25. The SLC35A2 gene opens another avenue for targeted treatment in MCD and is outside of the well-recognized mTOR pathway associated with FCD ILAE Type 2 lesions15. A recent report showed a clinical improvement in response to oral D‐galactose supplementation in several SLC35A2-congenital disorder of glycosylation (CDG) patients, caused by monoallelic pathogenic variants in SLC35A2 on chromosome Xp11.23, resulting in reduction of seizure frequency 32. Evaluating this therapeutic approach in patients with brain mosaic mutations of SLC35A2 and MOGHE, and for whom surgery is not an option or failed to reduce seizures due to the large extent of the lesion, could offer a personalized treatment strategy. Yet, there is no targeted animal model of SLC35A2 brain mosaicism to clarify the precise pathogenicity. It also remains to be shown how a primary defect in glycosylation in neuroglial progenitors leads to the histopathological features described in MOGHE, i.e. patchy oligodendroglial hyperplasia and heterotopic neurons in the white matter (Figure 4).
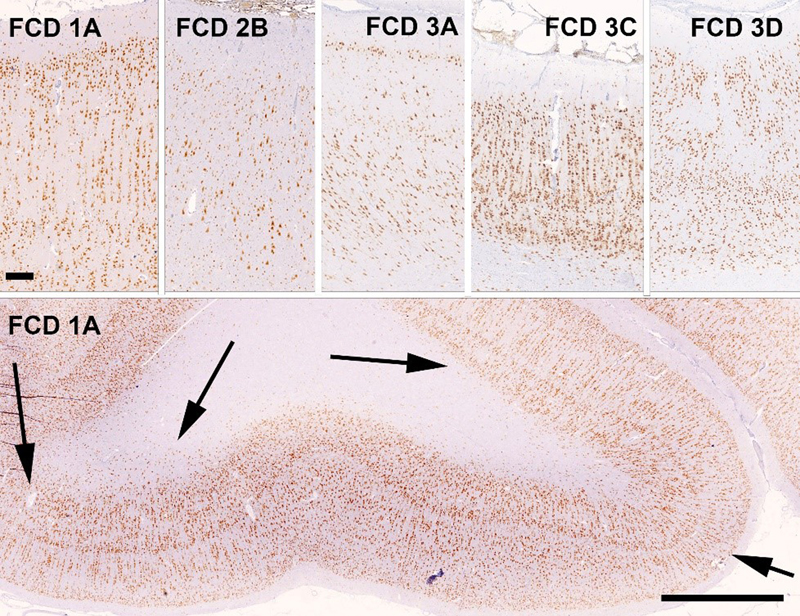
Figure 4: Simplified representation of pathophysiology and neuropathology findings in SLC35A2-related MOGHE cases On the left: SLC35A2 loss-of-function variants cause a defect in protein/sphingolipid glycosylation in the cell. On the right: Loss of SLC35A2 function leads to a MOGHE phenotype, with clusters of increased density of Olig2-positive cells in the white matter and deep cortical layers. L1-L6: layers 1-6; WM: white matter; Figure taken from 29, an open access article distributed under the terms of the Creative Commons CC BY license. In 2021, another comprehensive study described the clinico-pathological and molecular presentation of FCD ILAE Type 1A33. Holthausen identified a cohort of 19 young children with drug-resistance from seizure onset and severe EEG abnormalities almost always emphasized in the posterior (temporo-parieto-occipital) quadrant. The lack of focal neurological deficits but severe cognitive impairment was another hallmark of Holthausen’s disease. Important for the diagnostic work-up is the MRI signature presenting as subtle hypoplasia of the epileptogenic area with focus on the white matter. Abundance of cortical microcolumns, best visible by immunohistochemical stainings using NeuN antibodies and detected in all 19 cases assigned Holthausen’s disease to FCD ILAE Type 1A (Figure 5), molecularly confirmed by DNA methylation analysis. These defining features coined the provisional term “multilobar unilateral hypoplasia with severe epilepsy in children (MUHSEC) and is referred to Holthausen’s disease herein. To the best of my knowledge, this case series is a first comprehensive electro-clinical and anatomo-pathological description of a pure FCD ILAE Type 1 cohort in the contemporary peer reviewed literature. The three FCD1 subtypes have been almost hypothetically defined by the ILAE classification scheme as architectural abnormality of the 6-layered neocortex without frank signs of cytopathology, i.e., dysmorphic neurons or balloon cells. Up to date, there is no clear description available for either FCD 1B (horizontal dyslamination) or FCD 1C (horizontal and vertical dyslamination), although the latter is often associated with developmental vascular lesions, e.g., meningeal angiomatosis of Sturge-Weber34, or perinatal stroke, and assigned to FCD 3C or 3D, respectively (Figure 5)6,35. There was always concern that different patterns of cortical dyslamination are caused by different aetiologies and pathophysiological mechanisms, which will have implications for the diagnostic work-up as well as for the therapeutic management36. Yet, we have to await further studies until the issue of FCD Type 1 subtypes and their clinical relevance for patient management and treatment options will be finally resolved.
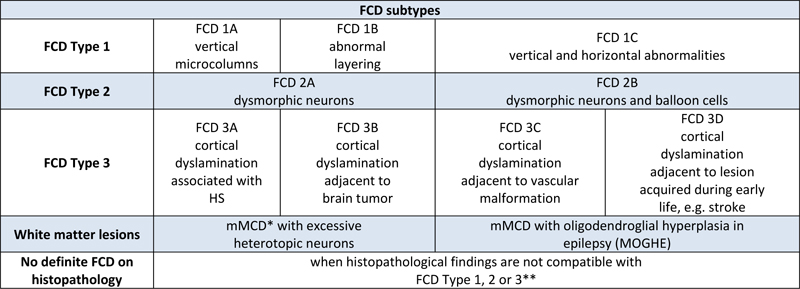
Figure 5: FCD subtypes of the ILAE classification scheme of 2011 as visualized with NeuN immunohistochemistry Upper Panel with the pial surface always presented on top. NeuN immunohistochemistry with hematoxylin counterstain. Scale bar on upper left = 250 μm, applies to all images of upper panel. Scale bar on lower panel = 2500 μm. FCD1A: 2-year old girl with right temporo-parieto-occipital seizures. The histograph reveals abundance of microcolumns. This is even more visible on the low-power magnification of the same patient in the lower panel with arrows indicating several large regions with microcolumns and heterotopic neurons in the white matter. FCD 2B: 5-year old boy with left frontal lobe epilepsy. Note the large (dysmorphic) neurons in an otherwise unlayered neocortex. FCD 3A: 28-year old male patient with left-sided temporal lobe epilepsy and hippocampal sclerosis (not shown). Note the pathognomonic narrowing of cortical layer II and depletion of pyramidal neurons in layer III. FCD3C: 20-year old male patient with Sturge-Weber syndrome. Note the meningeal angiomatosis on top and the abundant microcolumnar arrangement of neurons in the neocortex. Adjacent cortical areas also revealed disorganization of cortical layers in a horizontal direction (although not visible by this magnification). FCD3D: 14 year old girl with seizures and intrauterine stroke. Note the architectural abnormalities in cortical layers II-V. TOPIC 5: Toward a first update of the international consensus classification of FCD FCD are the most common malformations of cortical development recognized in epilepsy surgery case series2,13. Clinical diagnosis is usually based on the presurgical evaluation including high- or ultra-high-resolution MRI37, but should be always confirmed histopathologically from surgically resected brain samples. Hence, the agreement of microscopic diagnoses remains ever challenging, which has been documented many times in the scientific literature3-7. In 2021, a new approach was launched by a consortium nominated by the ILAE Task Force of FCD. They iteratively reviewed a consecutive cases series of 22 patients from a single center in the United States and with each patient being clinically (presurgically) suspicious for any type of FCD6. In addition, five independent laboratories performed genetic testing with FCD-specific gene panel sequencing from tissue and blood samples of the very same 22 patients. Four rounds of histopathology agreement were performed amongst 20 internationally renowned colleagues from 16 countries. The group started with a series of 196 H&E stains made available through an open access digital slide review platform. Notably, agreement was very low (kappa value = 0.16), and all reviewers were also asked to propose useful immunohistochemical stains from sections with the most prominent histopathologic changes. Antibodies directed against the NeuN, MAP2 and non-phosphorylated neurofilament epitopes were requested most often and can thus be considered as most valuable for the histopathology work-up of epilepsy surgery specimens6. Notwithstanding, the panel of recommended immunostainings cover many more epitopes, which may become necessary in the differential diagnosis of difficult-to-classify epileptogenic brain lesions38. This panel has been recently extended including mutation-specific antibodies directed against BRAFV600E and IDH1R132H, as well as the pS6 epitope to visualize an activated mTOR pathway (Najm et al. The ILAE FCD classification update 2022, in revision). With the addition of requested immunohistochemical stains the agreement increased to a kappa-value of 0.35 in the second round. During the third round, all previous answers were anonymously disclosed using the Delphi consensus method. MRI and histographs showing specific features of each lesion were also disclosed for the consensus discussion. This increased the diagnostic agreement to a kappa value of 0.5. In the final round, the results from genetic testing were available, i.e., pathogenic MTOR, DEPDC5, AKT3, NPRL3 and SLC35A2 mutations in 7 cases. The final agreement based on all available information was substantial with a kappa value of 0.686. This study will define the path towards an integrated genotype-phenotype diagnosis in FCD and the authors proposed a number of amendments to be recognized in an update of the 2011 ILAE classification scheme (Table 1). Most importantly, the authors suggested to include two new entities, which were predominantly related to the white matter and already described in the scientific literature, but not yet well recognized in the neuropathology work-up of epilepsy surgery specimens. The MOGHE entity has been already discussed above and the association with pathogenic SLC35A2 brain somatic mutations was confirmed in the presented case series. Yet there is no hotspot mutation nor any immunohistochemical surrogate marker available for SLC35A2. The other entity is that of mMCD with abundance of heterotopic neurons in the white matter as a defining hallmark. This lesion type will surely remain a matter of concern and discussion, as few heterotopic neurons are also present in the normal white matter of the non-epileptic cortex, as well as in several other epileptogenic disease conditions, first and foremost in FCD ILAE Type 1A (Holthausen’s disease) and MOGHE. In addition, the term mMCD should be used only when any other pathology finding was ruled out, including any other principal lesion such as hippocampal sclerosis, developmental brain tumors or glial scars acquired early during life. The authors further suggested to include a panel of immunohistochemical markers to confirm the diagnosis of FCD or when no lesion can be microscopically defined6.
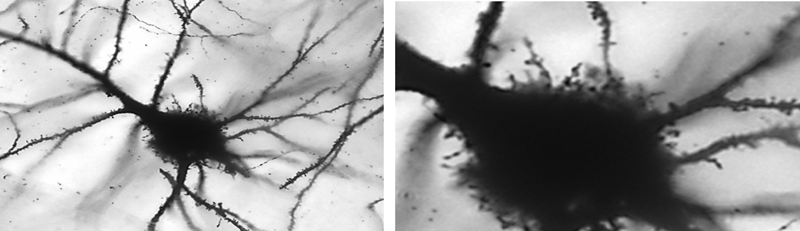
Table 1: Proposed amendments to the ILAE classification scheme of FCD FCD, Focal Cortical Dysplasia, HS, Hippocampal Sclerosis, mMCD, mild malformation of cortical development, MOGHE, mMCD with oligodendroglial hyperplasia in epilepsy; * mMCD: not associated with any other principal lesion, such as hippocampal sclerosis, brain tumor, or vascular malformation; for reference values see39-42. ** No definite FCD on histopathology: a descriptive report is recommended to highlight anatomical ambiguities (if applicable). Table modified from 6, an open access article distributed under the terms of the Creative Commons CC BY license. The proposed amendments to the classification scheme presented in Table 1 were taken from the discussed publication6, whereas the actual ILAE classification update is still under peer review and expected to be published in due course of 2022 (Najm et al. The ILAE FCD classification update 2022, in revision). This FCD classification update will propose for the first time a multi-layered integration of all available diagnostic information, including histopathology (Layer 1), genetics (Layer 2) and imaging (Layer 3), in order to achieve a final diagnosis. This approach may also compensate for the lack of accuracy or agreement shown for each diagnostic layer on its own, due to the aforementioned issues and challenges in the histopathology assessment6. It will then be the task of the treating physician to assemble all these data to a final genotype-phenotype diagnosis. A likely scenario to foster and promote this final diagnosis would be a post-surgical patient management conference, including the epileptologists, neurosurgeon, neuropathologist, neuroradiologist and neurogeneticist. The proposition to add the level of genetic testing to a comprehensive, reliable and integrative genotype-phenotype diagnosis represents an important step towards precision medicine in the realm of epileptology, as has been successfully implemented in neuro-oncology. Genetic testing should ideally be performed from DNA extracted from a microscopically assessed surgical brain tissue and paired blood sample, to facilitate the identification of brain mosaicism that represent post-zygotic mutational events. Whether or not such genetic testing should become mandatory in clinical practise or should be applied foremost in scientific research will be a matter of further discussion and will always depend on available resources in each epilepsy (surgery) centre. The possibility to detect brain somatic mutations in cell-free DNA obtained from cerebrospinal fluid using targeted droplet digital PCR was discovered in 2021 and described by two papers18,19. These findings offer the opportunity to establish a genetic diagnosis even before surgery, or in patients not eligible for surgery, and to guide the implementation of targeted therapies into the arena of epileptology. TOPIC 6: News from low-grade epilepsy-associated brain tumors (LEAT): human studies The fifth edition of the WHO classification of brain tumors is now available online and in print43. This 5th edition introduced six new entities pertinent to the arena of epileptology (out of 22 overall new tumor types, 27%), namely the many variants and subtypes of glio-neuronal tumors and low-grade gliomas, i.e., paediatric type diffuse low-grade gliomas, MAPK pathway-altered; diffuse astrocytoma, MYB or MYBL-altered; polymorphous low-grade neuroepithelial tumor of the young; diffuse glioneuronal tumor with oligodendroglioma-features and nuclear clusters; myxoid glioneuronal tumor; and multinodular and vacuolating neuronal tumor. Their molecular-pathologic features have been scientifically defined but the epileptological disease condition with early seizure onset and drug-resistant epilepsy as clinical hallmarks and their long-term seizure outcome following gross surgical resection were rarely addressed at a comprehensive level. As a result, most of the new tumor entities are not yet introduced to the clinical arena of epileptologists and a careful review of published histopathology images in the new WHO classification suggests that these new entities mimic well-established tumors of the LEAT family (LEAT – low-grade and epilepsy-associated tumors)43. One example is that of diffuse astrocytoma, MYB- or MYBL1-altered, which was first described in 2004 as isomorphic astrocytoma in a series of 19 patients with long-term and drug-resistant epilepsy44,45 or the polymorphous low-grade neuroepithelial tumor of the young (PLNTY), which shares many structural (i.e., glio-neuronal) and molecular (i.e., CD34, BRAF V600E) similarities with gangliogliomas (GG)43,46. A recently published genotype-phenotype study from Beijing, China addressing 30 patients with GG confirmed the presence of BRAF V600E in the majority of their GG cases (77%) when submitted to targeted next-generation sequencing using a panel of 131 genes47. There was no low-grade paediatric type diffuse glioma, PLNTY, nor any other of the new subtypes specified above present in their series. Along these lines, a group of French neuropathologists have asked the question whether the staggering number of new LEAT entities make a difference in the clinical management of epilepsy48. They focused on a subgroup of 72 LEAT cases, predominantly presenting with an oligodendroglial phenotype, and tried to classify them according to the 2021 WHO classification scheme. Based on RNA-sequencing, multiplexed digital PCR, DNA methylation analysis and histopathology review they identified only two major clusters of biological significance, with cluster 1 being enriched with dysembryoplastic neuroepithelial tumors (DNT) at histology, belonging to the LGG-DNT methylation class with CD34 negativity and FGRF1 alterations; while cluster 2 was classified histopathologically as GG, belonging to the LGG-GG methylation class, with BRAF V600E mutation and CD34 positivity. The same conclusion was already drawn in 2019 by an international LEAT review panel of 25 experts from 18 countries when studying their series of 30 LEAT7. The current situation questions, therefore, the clinico-pathological relevance of the various types recognized by the WHO within the spectrum of LEAT48. It will remain a continuous debate and negotiation to bring together all experts, including histopathologists, epileptologists and neuro-oncologists, to define a successful strategy for best patient management. These tumors rarely progress into malignancy but offer the opportunity for long-term seizure freedom when neurosurgically resected following a comprehensive epileptology work-up2. This will require new measures and parameters for the definition of LEAT when addressing the specific needs of patients with early onset drug-resistant epilepsy. TOPIC 7: News from LEAT: animal models BRAF V600E is the most common genetic driver in LEAT and is now variably assigned to several tumor entities listed in the 2021 WHO classification update (see above). Notwithstanding, none of these entities share an easy to classify histopathology phenotype and the failure to establish and agree upon the diagnosis across countries and laboratories is well known from published literature49. Intuitively, one would expect that a gene driver mutation such as BRAF V600E would establish a single diagnostic entity rather than being implicated in separate neuroepithelial tumors, i.e., paediatric type diffuse low-grade gliomas, MAPK pathway-altered; PLNTY; pleomorphic xanthoastrocytoma; GG, DNT, or multinodular and vacuolating neuronal tumor. When BRAF V600E is experimentally introduced in utero into the developing murine brain by intraventricular electroporation, the resulting phenotype was consistently described as biphasic GG by Koh et al. in 201850. The resulting lesion also showed intrinsic epileptogenic properties in the neuronal cell lineage, whereas tumorigenic properties were attributed to the proliferation active glial cell lineage. RNA sequencing analysis of patients’ brain tissues with the mutation revealed that BRAF V600E-induced epileptogenesis is mediated by RE1-silencing transcription factor (REST), which is a regulator of ion channels and neurotransmitter receptors associated with epilepsy50. Moreover, they found that seizures were treatable in mice with the FDA-approved BRAF V600E inhibitor vemurafenib, as well as various genetic inhibitors of REST. Accordingly, this study provides direct evidence of a BRAF somatic mutation contributing to the intrinsic epileptogenicity in paediatric brain tumors and suggests that BRAF and REST could be treatment targets for intractable epilepsy. Cases-Cunillera and co-worker have used the same technique of in utero electroporation to extend these studies towards BRAF V600E mutations with double- and triple-hits51. In their most recent animal model, aberrant BRAF expression in murine neural progenitors generated benign glio-neuronal neoplasms of the GG phenotype only in concert with active mTOR-signaling, i.e. by phosphorylated Akt (pAkt). Additional somatic Trp53-loss generated anaplastic GG, a grading scale and terminology not supported anymore by the current WHO classification scheme. Functionally, only BRAF/pAkt altered tumors showed substantial neuronal activity which spread to the adjacent neocortical tissue, which was interpreted as correlate of increased epileptogenicity51. In both animal studies, however, CD34 immunoreactivity was an immunohistochemical surrogate of the experimentally induced GG50,51. Such experimental LEAT models, which reproduce the variable genetic and histopathologic tumor phenotypes, will be important tools to further our knowledge and finally assess targeted therapies. TOPIC 8. What is new about hippocampal sclerosis?: Human Herpes Virus infection and the impact of inflammatory infiltrates on neuronal cell loss Hippocampal sclerosis (HS) remains the single most common cause of focal epilepsy amenable to epilepsy surgery13. Its etiology remains, however, to be further clarified with most researchers probably agreeing with the statement that HS is an end stage resulting from multiple etiologies. This may also be reflected by the different histopathologic patterns of segmental neuronal cell loss in HS being recognized in the ILAE consensus classification scheme of 201352. However, inflammation remains a major topic of interest in the pathogenesis of mesial temporal lobe epilepsy (MTLE) with or without HS. I will briefly discuss three publications highlighting this topic. Tröscher and colleagues analyzed the presence of T cells in various MTLE subgroups to answer the question of how much inflammation is present and whether the presence of T cells is associated with seizures and/or neuronal cell loss in the hippocampus53. Their detailed histopathological investigation of the involvement of T cells in various MTLE subgroups, i.e., gangliogliomas, febrile seizures, postinfectious encephalitis and Rasmussen encephalitis w/o HS, suggested that T cell numbers correlated with the degree of neuronal cell loss rather than seizure frequency or disease duration. Their quantification also showed that T cell numbers were significantly elevated in all MTLE groups compared to healthy post-mortem controls. However, CD3+ as well as CD8+ T cell numbers varied among their MTLE subgroups and nearly all MTLE groups revealed elevated numbers of T cells years after the precipitating injury. Hippocampal infection with Human Herpes Virus 6 (HHV-6) has been demonstrated in patients with MTLE already in 200354 and confirmed thereafter in several scientific publications. It can be envisioned, therefore, as one key etiological cause of MTLE with HS. This knowledge gains further momentum from the publication of Theodore and coworkers published in 2021, detecting HHV-6A and HHV-6B strains in fresh human tissue samples obtained from 87 patients with drug-resistant epilepsy submitted to epilepsy surgery55. Twenty-nine of their 54 patients with HS (54%), six of 23 with FCD (26%), and one of three with a history of encephalitis (33%) were positive for HHV-6. This contrasts the overall low percentage of HHV-6 DNA detection in only 6% of human brain samples with non-neurologic illness56. A febrile seizure history was not significantly associated with HHV-6 detection. However, patients with HHV-6 positive HS had significantly lower age at seizure onset than those with other pathologies. Also, there was a trend for HHV-6 positive patients to have higher binding of [11C]PBR28 in positron emission tomography (PET), the latter being a marker for reactive astrocytes and activated microglia, suggesting an inflammatory pathomechanism. Indeed, these findings reinforce the potential role and impact for HHV-6 in the etiology of MTLE with HS. However, the same group of researchers published a neuroimaging approach in 2020 to address the effects of HHV-6 on hippocampal volume in patients with hippocampal sclerosis57. They used MRI post-processing to segment cortical structures and to obtain an asymmetry index between hippocampal volumes ipsilateral and contralateral to the seizure focus when comparing between HHV-6 positive and negative patients. In this study, however, HHV-6 negative patients had significantly greater asymmetry and lower total hippocampal volume ipsilateral to the seizure focus compared to HHV-6 positive patients. Disease duration and age of onset did not affect these results. Their data suggest that HHV-6 still play a role in MTLE but with HHV-6 having a less severe effect on hippocampal damage. This controversy will be ongoing, however, and in need of more studies to clarify the issue. TOPIC 9. The fine structure of the epileptogenic neocortex and white matter Gray-white matter blurring of the anterior temporal lobe (GWMB) is a common neuroimaging finding in patients with HS58. It is often assigned to FCD, i.e., FCD ILAE Type 1, although systematic correlation studies of in vivo and ex vivo MRI with histopathology showed disturbed axonal myelination of the affected white matter as an underlying structural correlate rather than any signature of cortical (neuronal) malformation58. Whether or not such white matter alterations were due to secondary axonal degeneration, e.g., neuronal cell loss in the ipsilateral hippocampus, has yet to be clarified but was already suggested as severity aggravates with disease duration58. This topic was once again addressed in a study by Demrath and co-worker in 2021, when studying MRI-histopathology correlations in twenty patients with unilateral temporal lobe epilepsy, GWMB and HS59. Anterior temporal lobe white matter T1 relaxation times and diffusion measures were analyzed on the side of HS, on the side contralateral to HS, and in ten normal controls. Resected brain tissue was further evaluated at the ultrastructural level from three patients without GWMB and four patients with GWMB addressing axon density and diameter, the relation of the axon diameter to the total fiber diameter, and the thickness of the myelin sheath59. As a result, HS with GWMB of the anterior temporal lobe was related to prolonged T1 relaxation and axonal loss. A less pronounced reduction in axonal fraction was also found on imaging in GWMB-negative temporal poles compared to normal controls. Contralateral values did not differ significantly between patients and normal controls. Reduced axonal density and axonal diameter were histopathologically confirmed in samples with GWMB compared to temporal poles without GWMB. The authors concluded that GWMB should be considered as an imaging correlate for disturbed axonal maturation that can be quantified with advanced diffusion imaging. In my view, however, the data does not provide true evidence for “disturbed axonal maturation” in the sense of a maldevelopmental feature. Similar findings may occur as secondary retrograde damage and/or repair of the axonal compartment as suggested previously58. In 2021, another study addressed the fine microstructure of the epileptic neocortex using capricious Golgi-impregnations to further analyze synaptic networks of glutamatergic and GABAergic axon terminals60. Alterations in dendritic morphology and spine loss have been reported both in epilepsy animal models and in human brain tissues from patients with epilepsy61. However, it is still unclear whether these dendritic abnormalities relate to the cause of epilepsy or are generated by seizure recurrence. Rossini and coworker investigated these fine neuronal structures in cortical specimens from 28 patients with different neuropathologically defined etiologies, i.e., FCD Type 1A and Type 2, and non-lesional neocortex obtained from TLE with HS. Autoptic brain tissues were used for comparison. Three-dimensional reconstructions of Golgi-impregnated neurons revealed severe dendritic reshaping and spine alteration in the core region of FCD Type 2. Dysmorphic neurons showed increased dendritic complexity, reduction of dendritic spines and occasional filopodia-like protrusions emerging from the soma (Figure 6). Surprisingly, the intermingled normal-looking pyramidal neurons also showed severe spine loss and simplified dendritic arborization. No changes were observed outside the dysplasia in perilesional tissue or in neocortical tissue obtained from the other patient groups. These data confirmed a rather normal appearing fine morphological aspect of neurons and dendritic spines in the epileptogenic neocortex, with the exception of type 2 dysplastic lesions and argue against the concept that long-lasting epilepsy will produce per se any dendritic pathology.
Figure 6: Filopodia-like protrusions in dysmorphic neurons of FCD 2B Low power microphotographs of Golgi-impregnated dysmorphic neuron from a FCD 2B on the left, showing the presence of numerous short filopodia-like protrusions emerging from the soma. Higher magnification on right. Modified from 60 with permission from the author. The missing TOPIC 10: What is still missing to further the advancement of neuropathology and epilepsy surgery? We are already facing another major advancement in neuropathology, which will soon be recognized as the era of digital neuropathology. Artificial intelligence (AI)-based disease classifier and ever growing digital slide suites will substitute the routine work-up and may be able to also predict essential molecular features of the underlying disease condition to inform precision medicine treatment. These topics have not yet been publicized in peer reviewed journals with the exception of a deep learning-based algorithm to differentiate FCD Type 2 from cortical tuber on routine H&E-stained glass slides in 202062. AI-based algorithms may even be able to replicate histochemical and immunohistochemical stainings from unstained slides without actually performing the staining procedures63,64. This potential is yet to be fully explored, including within the field of neuropathology and epilepsy surgery, and we will await upcoming publications addressing this fascinating matter as it may well reshape our next future in neuropathology. Conclusion During the past decade, there have been considerable advances in understanding the genetic and morphogenic processes underlying cortical malformations and developmental brain tumors15. Focal brain malformations result from somatic (post-zygotic) variants in several genes related to the mTOR pathway in Focal Cortical Dysplasia Type 2, AKT3, DEPDC5, MTOR, NPRL2/3, PIK3CA, RHEB, TSC1/2 and others, or in the galactose transporter gene SLC35A2 in MOGHE, which were acquired early during cortical development. The timing of the genetic event, the specific gene involved and the targeted precursor cell population will determine the nature and size of the lesion, whether developmental malformation or a brain tumor. There is also emerging evidence of epigenetic processes underlying a ‘molecular memory’ in epileptogenesis22, which also helped to establish a diagnostic DNA methylation classification scheme21. This knowledge will finally accumulate into a better understanding of why and how patients with these lesions have epilepsy and to move toward precision medicine in patients with drug-resistant focal epilepsy. These topics will be continuously addressed and discussed also at the annual ILAE summer school of Neuropathology and Epilepsy Surgery (https://www.ilae.org/congresses). Please address your application to bluemcke[at]uk-erlangen.de References
Copyright: © 2022 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |