|
|
|
Free Neuropathology 2:24 (2021) |
|
Case Report |
|
The Multifaceted Appearance of Supratentorial Ependymoma with ZFTA-MAML2 Fusion |
|
Ming Liang Oon1, Lutfi Hendriansyah2, Patricia Diana Pratiseyo3, Eka J Wahjoepramono2, Jian Yuan Goh4, Chik Hong Kuick4, Kenneth TE Chang4,5, Arie Perry6, Char Loo Tan1,4 |
|
1 Department of Pathology, National University Health System, Singapore, Singapore |
|
Corresponding author: |
|
Submitted: 12 June 2021 Accepted: 11 September 2021 Copyedited by: Jerry J. Lou Published: 20 September 2021 |
|
Keywords: Ependymoma, ZFTA, C11orf95, MAML2, Myogenic |
|
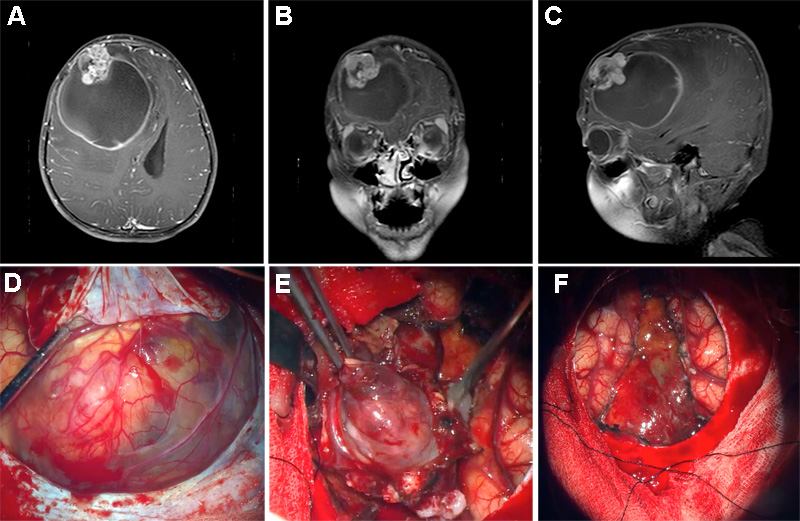
Abstract Ependymomas are glial neoplasms with a wide morphological spectrum. The majority of supratentorial ependymomas are known to harbor ZFTA fusions, most commonly to RELA. We present an unusual case of a 9-year-old boy with a supratentorial ependymoma harboring a noncanonical ZFTA-MAML2 fusion. This case had unusual histomorphological features lacking typical findings of ependymoma and bearing resemblance to a primitive neoplasm with focal, previously undescribed myogenic differentiation. We discuss the diagnostic pitfalls in this case and briefly review the histological features of ependymoma with noncanonical gene fusions. Our report underscores the importance of molecular testing in such cases to arrive at the correct diagnosis. Supratentorial ependymomas with noncanonical fusions are rare, and more studies are necessary for better risk stratification and identification of potential treatment targets. Introduction Ependymomas (EPN) form a diverse group of morphologically and molecularly heterogeneous glial neoplasms [5, 14]. While the classical histological appearance of EPN is well-described, the morphological spectrum is broad and may confound diagnosis. Unusual features encountered in ependymomas include chondro-osseous elements [13, 17], neuropil-like islands [6], melanin-containing cells [4], tumor giant cells [8], neuronal differentiation [16], granular cell features [18], et cetera. Recent advances in genomic and methylome profiling have identified distinct molecular subgroups characterized by recurrent genetic or epigenetic alterations that may better inform diagnosis, clinical outcomes and risk stratification of EPN as compared to conventional light microscopic diagnosis and histological grading [5]. The majority of supratentorial (ST) EPNs have gene fusions involving either ZFTA (zinc finger translocation associated; previously known as C11orf95) or YAP1. These gene fusions characterize the two molecular subgroups of ST-EPN: ST-EPN-ZFTA and ST-EPN-YAP1, respectively [1, 14]. Here, we present an unusual case of a ST-EPN with ZFTA-MAML2 fusion demonstrating diagnostic challenges due to primitive morphology, suggesting an embryonal neoplasm and divergent differentiation, including hitherto undescribed myogenic differentiation. Our case underscores the importance of molecular testing in such cases to arrive at the correct diagnosis. Clinical summary A 9-year-old boy presented with a visible, enlarging right forehead lump for 2 months. The child complained of an occasional headache but was otherwise well without any focal neurological deficits. Brain magnetic resonance imaging (MRI) revealed a well-demarcated, right frontal cystic tumor with a mural nodule, surrounding vasogenic edema and midline shift (Fig. 1A-C). The tumor showed a high choline peak and high choline:N-acetylaspartate (NAA) ratio. The child underwent craniotomy, revealing a well-demarcated bosselated tumor with a clear interface between tumor and brain (Fig. 1D-E). Gross total resection of the tumor was achieved (Fig. 1F), and the postoperative course was uneventful. He received adjuvant radiation therapy followed by temozolomide. The child remained well at follow up 9 months post-resection, with no radiological evidence of recurrence.
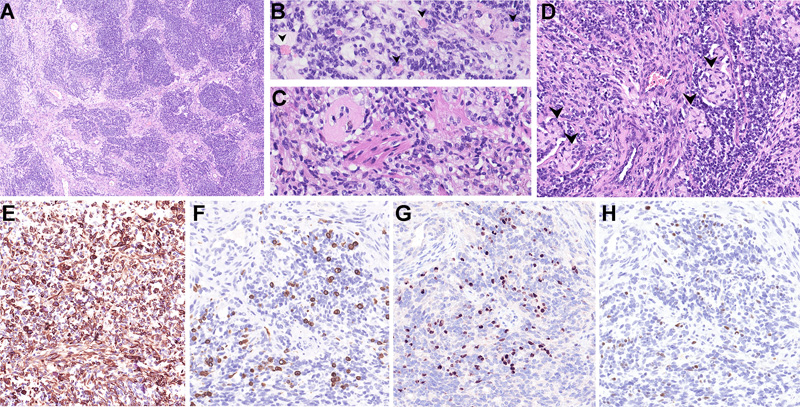
Figure 1. MRI findings of the tumor. A) Axial, B) coronal and C) sagittal views showing a cystic tumor with an enhancing multilobulated mural nodule in the right frontal lobe with mass effect. D) Intraoperative view after lifting of dural flap, prior to resection E) Dissection of a well-demarcated multinodular cystic tumor. F) Final intraoperative view after gross total resection of the solid tumor showing part of cyst lining with ventricular wall. Material and methods The specimen was routinely processed for paraffin embedding and staining with hematoxylin and eosin (H&E). Immunohistochemistry (IHC) with commercially available primary antibodies, including GFAP, OLIG2, synaptophysin, desmin, myogenin, MyoD1, CAM 5.2, EMA, INI1, BRG1, LIN28A, BCOR, HMB45 and Ki67, was performed following locally validated technical protocols. Gene fusion detection was performed using a next-generation sequencing-based anchored multiplex polymerase chain reaction (PCR) assay (Archer® FusionPlex, Boulder, CO, USA) that can detect and identify gene fusions involving any of 101 covered genes, including RELA, YAP1, MAML2, NCOA1 and NCOA2. Briefly, total ribonucleic acid (RNA) was extracted from formalin-fixed paraffin-embedded tissue sections of tumor. 150 ng of RNA was used for library preparation utilizing the Archer® FusionPlex kit, according to the manufacturer’s protocol (ArcherDX, Boulder, CO, USA). The prepared library was sequenced using an Illumina MiniSeq sequencer. The data obtained was analyzed by the Archer Data Analysis (version 6.2.3) portal. A confirmatory reverse transcription-PCR was designed flanking the breakpoint of the two genes and the PCR product was Sanger-sequenced. DNA methylation analysis was performed using the Illumina Infinium Human Methylation EPIC BeadChip Kit according to published protocols [2, 7]. The results of the methylation profiling and copy number variation analysis were obtained using an automated web-based deoxyribonucleic acid (DNA) methylation profiling program (molecularneuropathology.org) [12]. The case was also integrated onto the t-distributed Stochastic Neighbor Embedding (t-SNE) analysis in the German Cancer Research Center (DKFZ) database. Results Light microscopy Sections showed a discrete tumor composed of hypercellular sheets of small, round to ovoid cells with hyperchromatic nuclei and indistinct cell boundaries, imparting a primitive appearance to the tumor. In other areas, the tumor cells formed nodular aggregates, short fascicles and palisades/ trabeculae, separated by fibrous septa imparting a desmoplastic appearance (Fig. 2A). A minor subset of tumor cells showed prominent intracytoplasmic fine eosinophilic granules, some with a more globular appearance (Fig. 2B). A minor component of the tumor was composed of spindled cells with eosinophilic cytoplasm reminiscent of skeletal muscle fibers (Fig. 2C). Focally, aggregates of tumor cells with eccentric nuclei and mucinous cytoplasm were also identified (Fig. 2D). No other heterologous differentiation was seen. There was a distinct lack of a fibrillary background in the tumor. While some tumor cells appeared to congregate around large thick-walled vessels, no distinctive perivascular pseudorosettes were present. Geographic areas of necrosis accompanied by dystrophic calcification were present. The mitotic count reached up to 8 per 10 high power fields (3 per mm2).
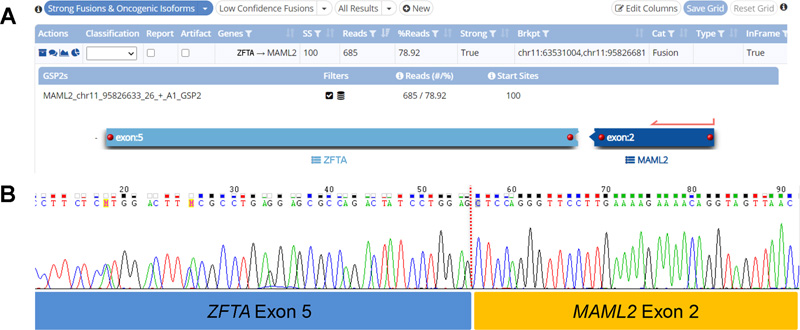
Figure 2. Light microscopy and immunohistochemical findings of the tumor. A) Low-power appearance of the tumor consisting of nests of tumor cells separated by fibrous septa, imparting a desmoplastic appearance. The bulk of the tumor cells show primitive cytomorphology with minimal cytoplasm, imparting a nondescript “small blue cell” appearance. No perivascular pseudorosettes are seen, although tumor cells aggregate along blood vessels. B) A minor subset of tumor cells showed prominent intracytoplasmic fine eosinophilic granules (arrowheads). C) Rare tumor cells showed spindled morphology, resembling skeletal muscle fibers. D) Aggregates of tumor cells with eccentric nuclei and mucinous cytoplasm (arrowheads). The tumor cells were positive for E) GFAP. Very focal areas showed myogenic differentiation, being positive for F) desmin, G) myogenin and H) MyoD1 (F-H were images taken from same area of the tumor.) Clicking the figure will lead you to the full virtual slide (H&E). The tumor cells showed patchy positivity for GFAP (Fig. 2E), and focal reactivity for synaptophysin, CAM 5.2 and EMA (cytoplasmic; no dot/ring-like pattern was found). Focally, some cells expressed desmin (Fig. 2F) and nuclear reactivity for myogenin (Fig. 2G) and MyoD1 (Fig. 2H). No neurofilament-positive axons were identified, in keeping with the non-infiltrative nature of this tumor. There was retained expression for INI1 and BRG1. OLIG2, LIN28A, BCOR and HMB45 stains were negative. The Ki67 labeling index reached up to 40%. Gene fusion detection and confirmation The Archer FusionPlex assay detected a ZFTA (exon 5)- MAML2 (exon 2) fusion (Fig. 3A). This gene fusion was confirmed by RT-PCR with Sanger sequencing of the amplicon product (Fig. 3B).
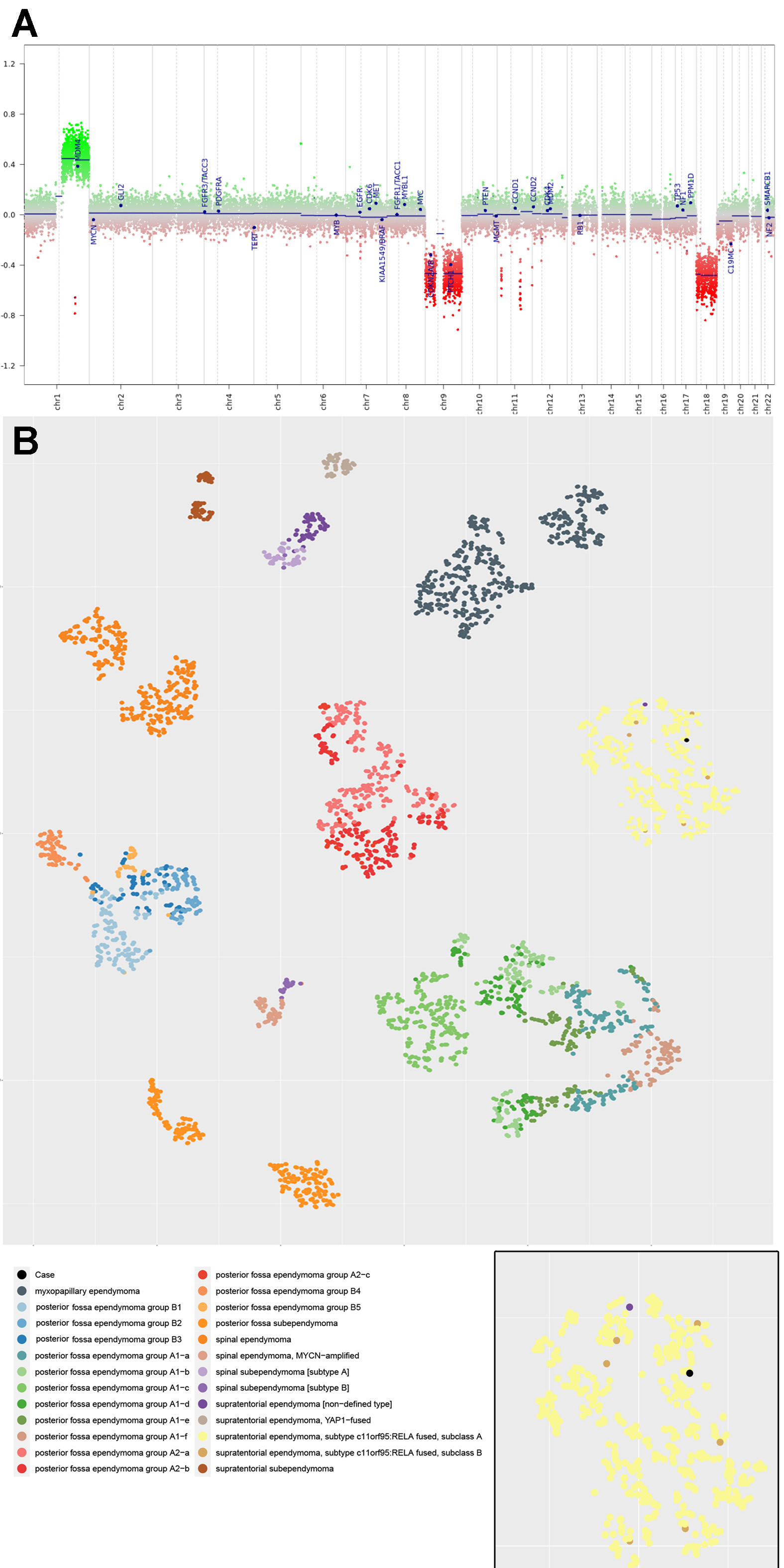
Figure 3. A) Archer FusionPlex assay detected a ZFTA (exon 5; previously known as C11orf95)-MAML2 (exon 2) fusion, which was confirmed by B) Sanger sequencing. Methylation analysis DNA-based methylation analysis of the tumor revealed a methylation class corresponding most closely to “Ependymoma, RELA fusion”, with a calibrated score of 0.65. Concurrent copy number analysis revealed 1q gain and CDKN2A/B loss (Fig. 4A). No chromothripsis of chromosome 11 was present. On the t-SNE plot, our case localized to the methylation cluster of ST-EPN-RELA, consisting of 444 cases including our case, out of a total of 3272 cases of ependymomas of different anatomical locations and molecular groups, lending further diagnostic confidence (Fig. 4B). A final diagnosis of ST-EPN with ZFTA-MAML2 fusion was rendered after synthesizing the pathological and molecular findings of this case.
Figure 4. A) Copy number variation profile of the tumor, with 1q gain and CDKN2A/B losses. B) t-distributed Stochastic Neighbor Embedding (t-SNE) plot containing a total of 3272 cases of EPNs for cluster analysis. Our case localized to methylation cluster of supratentorial EPN with RELA fusion (n= 444, including our case) (Inset, case indicated in black). Clicking the figure will lead you to a high-resolution version. Discussion ZFTA, now recognized as a key player in the oncogenesis of ST-EPN, confers transforming capabilities upon oncogenic fusion proteins by increasing their translocation to the nucleus and permitting access to the transcriptional machinery [9]. Most commonly, ZFTA is fused to RELA [9, 14, 19]. Being promiscuous, ZFTA may also rarely fuse with alternative partners such as MAML2, MAML3, NCOA1, NCOA2 and CTNNA2 [14, 18, 19]. Of note, the ZFTA-MAML2 fusion has only been identified in 18 cases of ST-EPN in the literature thus far, including the current case [14, 18, 19]. Tumors with such alternative fusion partners form satellite clusters around the previously known ST-EPN-RELA cluster in the t-SNE plot of methylation profiles and show distinct transcriptional profiles [19]. While most ST-EPN-ZFTA demonstrate classical features of EPN such as perivascular pseudorosettes and fibrillary matrix, a recent paper by Zheng et al found that EPNs with ZFTA fusions with alternative partners tend to demonstrate unusual histological features. These include sarcomatous features, areas mimicking high grade central nervous system (CNS) neoplasms like diffuse high-grade glioma, CNS embryonal tumors and other primitive tumors [19]. Such atypical histological findings are in keeping with the case we present herein. Similar to the case by Tamai et al, our case also demonstrated granular cell features [18]. In their case, the granular cells showed eccentrically placed nuclei and cytoplasm filled with eosinophilic granules which were positive for periodic acid-Schiff (PAS) and alpha-1-antitrypsin stains. In addition, our case showed a small population of tumor cells with unequivocal expression for desmin, myogenin and myoD1, consistent with myogenic differentiation, a finding which has not been previously described. Initial diagnosis on the basis of light microscopy was difficult because of the atypical morphologic features of the tumor including the conspicuous absence of a fibrillary stroma, the absence of classical EPN morphology, and the presence of primitive-appearing cells in a desmoplastic background. Coupled with the polyphenotypic immunoprofile, which included desmin reactivity, the differential of desmoplastic small round cell tumor was initially entertained [10]. However, the subsequent molecular finding of the ZFTA-MAML2 gene fusion and the absence of an EWSR1-WT1 fusion clinched the diagnosis of ST-EPN with ZFTA-MAML2 fusion and led to revision of the histologic diagnosis. Recurrent fusions involving ZFTA are characteristic of EPN and fusions involving this gene have only been described previously in chondroid lipoma [15]. No primitive tumor has been described in the literature with this characteristic fusion to date. While lacking in histological elements of classical EPN, the circumscribed, non-infiltrative nature of this tumor and the presence of the signature fusion and methylation profile were all in keeping with a diagnosis of ST-EPN. The novel finding of myogenic differentiation, likely attributable to metaplasia of the neoplastic glial component or the mesenchymal component of the tumor cells, broadens the spectrum of heterologous differentiation in EPN [3]. The protean morphological manifestations of EPN has the potential to obfuscate morphological diagnosis, particularly when classical EPN features are not present. This underscores the limitations of morphological evaluation alone and the necessity of molecular testing. There has been longstanding controversy surrounding the utility of histological grading of EPN. While ST-EPN-RELA was found to be associated with adverse patient outcomes in one retrospective study [14], this was not reproduced in another trial-based study [11]. Interestingly, mice with EPN induced by ZFTA-MAML2 fusion demonstrated a reduced survival compared to those with ZFTA-RELA fusion [19]. Out of the 18 cases of ST-EPN with ZFTA-MAML2 fusion, follow up data was only available in two cases. Both cases showed anaplastic features, corresponded to WHO grade 3. The case reported by Tamai et al. showed no recurrence for 30 months without chemoradiotherapy, which may indicate a favorable prognosis [18]. Our patient remained disease free at 9 months follow up. Owing to the paucity of survival data, it remains to be seen if differences in survival exist in ST-EPN with alternative fusions. Similarly, the utility of assignment of a WHO grade to molecularly defined EPN remains insufficiently characterized [5]. Additional studies are necessary to better delineate the behavior of these tumors, which may inform treatment decisions and risk stratification. In conclusion, we present an unusual case of ST-EPN with ZFTA-MAML2 fusion exhibiting primitive morphology and a focal area of previously undescribed myogenic differentiation. Awareness of the multifaceted appearance of EPN is critical and an integrated clinico-pathological-molecular diagnosis is essential for the contemporary diagnosis of this entity. Acknowledgements We would like to thank Drs. Felix Hinz and Felix Sahm from the German Cancer Research Center (DKFZ) for their invaluable help in getting the t-SNE data of our case. References
Copyright: © 2021 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |