|
|
|
Free Neuropathology 2:16 (2021) |
|
Case Report |
|
Molecular clarification of brainstem astroblastoma with EWSR1-BEND2 fusion in a 38-year-old man |
|
Matthew A. Smith-Cohn,1,2 Zied Abdullaev,3 Kenneth D. Aldape,3 Martha Quezado,3 Marc K. Rosenblum,4 Chad M. Vanderbilt,4 Fausto J. Rodriguez,5 John Laterra,**2 Charles G. Eberhart**5 |
|
1 Neuro-Oncology Branch, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA |
|
Corresponding author: |
|
Submitted: 21 April 2021 Accepted: 17 June 2021 Copyedited by: Calixto-Hope Lucas Published: 21 June 2021 |
|
Keywords: Astroblastoma, Brainstem, Fusion, Methylation, Neoplasms, Neuro-oncology |
|
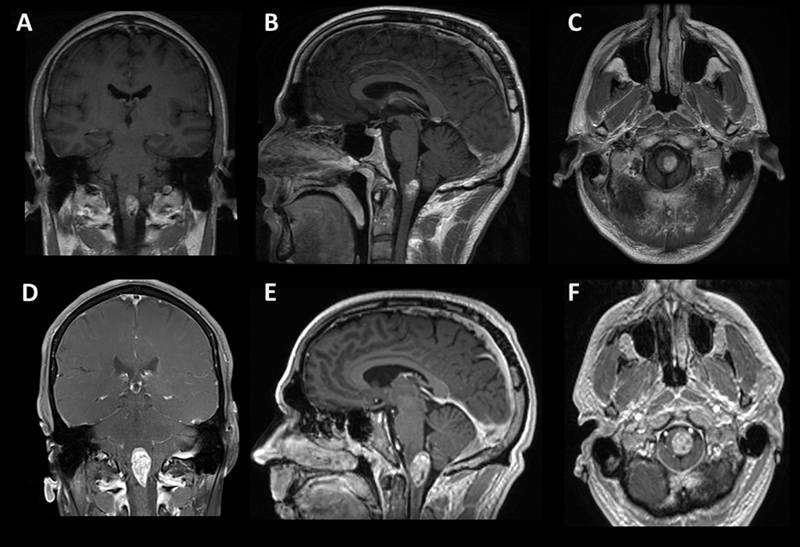
Abstract The majority of astroblastoma occur in a cerebral location in children and young adults. Here we describe the unusual case of a 38-year-old man found to have a rapidly growing cystic enhancing circumscribed brainstem tumor with high grade histopathology classified as astroblastoma, MN1-altered by methylome profiling. He was treated with chemoradiation and temozolomide followed by adjuvant temozolomide without progression to date over one year from treatment initiation. Astroblastoma most frequently contain a MN1-BEND2 fusion, while in this case a rare EWSR1-BEND2 fusion was identified. Only a few such fusions have been reported, mostly in the brainstem and spinal cord, and they suggest that BEND2, rather than MN1, may have a more critical functional role, at least in these regions. This unusual clinical scenario exemplifies the utility of methylome profiling and assessment of gene fusions in tumors of the central nervous system. Introduction Astroblastomas are rare central nervous system (CNS) neoplasms that most frequently occur in cerebral locations in children and young adults. Here we describe the unusual case of a 38-year-old man with a brainstem tumor with an integrated diagnosis of malignant neoplasm consistent with astroblastoma with MN1 alteration following methylome profiling, and found to have a non-canonical EWSR1-BEND2 fusion. This unusual clinical scenario exemplifies the utility of methylome profiling and assessment of gene fusions in tumors of the CNS. Case Report A 38-year-old male with a past medical history of melanoma in situ of the trunk, status post excision, and no family history of cancer, presented with subacute onset of progressively worsening dysesthesia first involving his upper extremities and progressing to affect his right chest and leg over two weeks. MRI of the brain and cervical spine demonstrated a well-localized cystic enhancing anterior medullary-cervical lesion measuring 1.3 x 1.1 x 1.7 cm, with T2 hyperintensity of the lesion and medullary pyramids (Fig.1A, 1B, 1C). He was admitted to the neurology service for an expedited evaluation of infectious, rheumatologic, neoplastic, and inflammatory etiologies. MRI of the spine and whole-body PET/CT revealed no other lesions. Serum studies were unremarkable, and lumbar CSF contained 1 WBC, 3 RBC, glucose 55, protein 57 (ref 15-45), no oligoclonal bands and negative infectious studies, flow cytometry, and cytopathology. He was treated with high dose IV methylprednisolone for five days and discharged home on a steroid taper. He had progression of symptoms, and a repeat MRI two months later showed growth of the lesion to 3.3 x 1.7 x 1.5 cm (Fig.1D, 1E, 1F). Subsequently, he underwent a suboccipital C1 laminectomy and subtotal surgical resection of the mass.
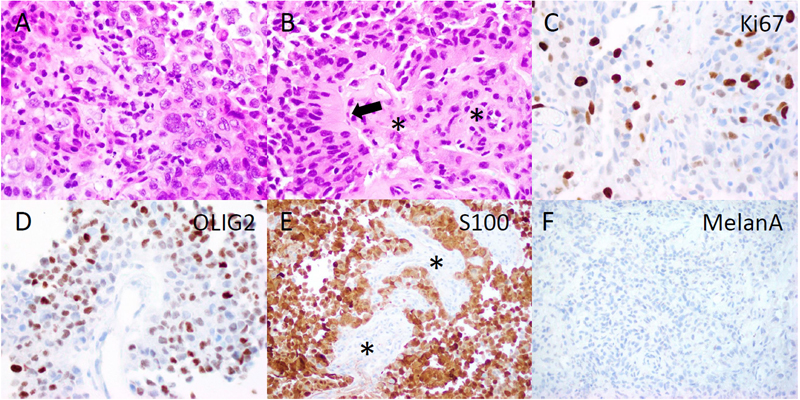
Figure 1. Radiographic findings. Coronal (A, D), sagittal (B, E), axial (C, F) post-contrast T1 MRI of the brain. The top row (A-C) shows MRI imaging of the brain at presentation, and the bottom row (D-F) is two months later before tumor resection. Pathology Microscopic evaluation showed a cellular tumor with compact growth. Large, pleomorphic to epithelioid cells predominated in some regions (Fig.2A), while in other areas prominent perivascular growth was noted. This included scattered cells with a somewhat astroblastic phenotype, exhibiting stout processes extending to the surface of blood vessels (Fig.2B). Necrotic foci without pseudopalisading were present. Cellular regions of tumor had 1 to 5 mitotic figures per high power field, and the Ki67 proliferation index was moderate to high, up to 20-30% (Fig.2C). S100, OLIG2 and EMA were strongly positive on immunohistochemical analysis (Fig.2D, E and data not shown), and GFAP was focally positive, supporting glial differentiation. In contrast, markers of melanocytic (MelanA, SOX10, HMB45), epithelial (cytokeratin AE1/AE3), and neuronal (synaptophysin) differentiation were all negative (Fig.2F and data not shown).
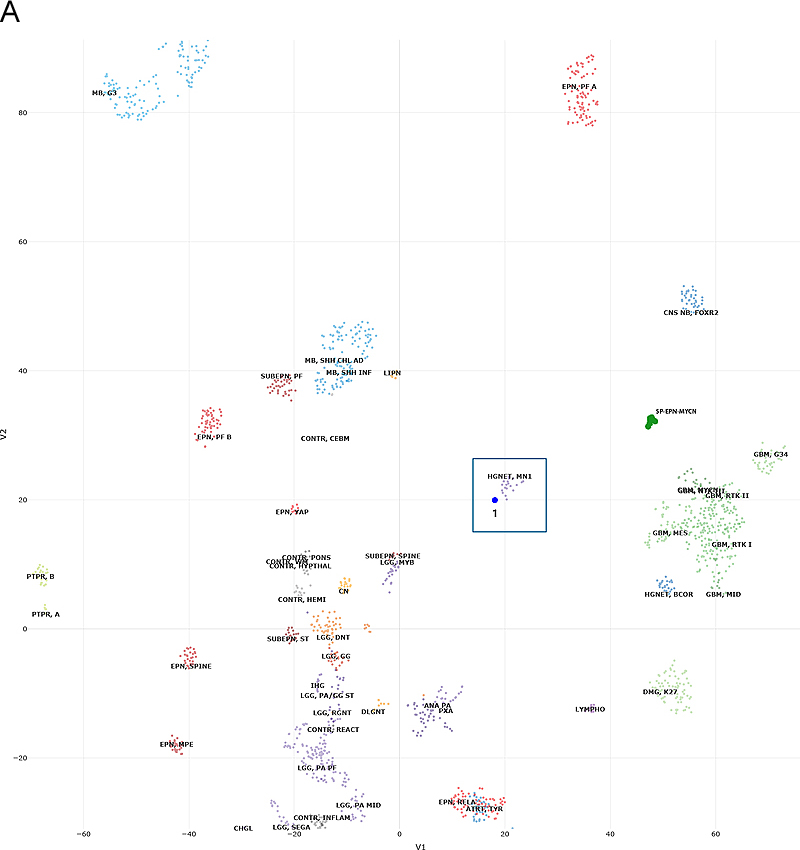
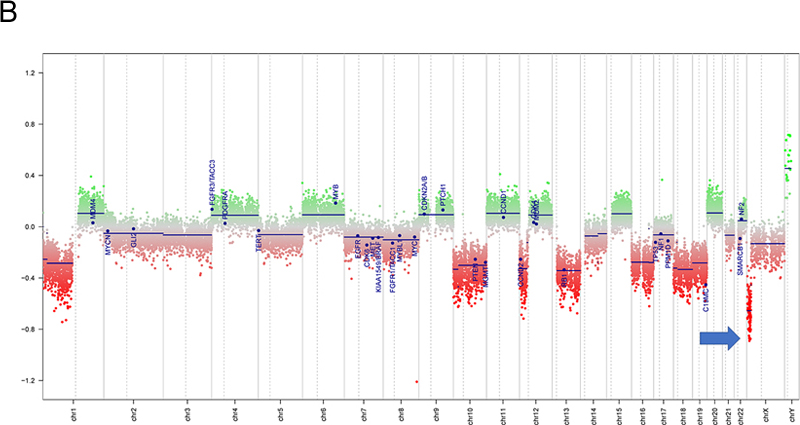
Figure 2. Histopathology. The tumor included more pleomorphic regions (A), as well as neoplastic cells with thick, short processes (arrow, B) arrayed around proliferating blood vessels (asterisks, B). The tumor was quite proliferative on Ki67 immunostain (C), and diffusely positive for OLIG2 (D) and S100 (E), while blood vessels did not express these glial markers (asterisks, E). MelanA was not expressed in the tumor (F). (Original magnifications: A-E 400X, F 200X). Molecular Diagnostics Next-generation sequencing (NGS) found a tumor mutation rate of 0.88 mutations per megabase, no known pathologic variants, and several variants of unknown significance, including ADGRA2 (p.G407S), AR (p.A646D), BLM (p.R643H), EPHA5 (p.L907V), and PRKDC (p.I1013V), all with allele frequencies of 46% or higher. No alterations in TP53, ATRX, BRAF, H3F3A or HIST1H3B were detected on NGS. DNA methylation profiling was consistent with a “high-grade neuroepithelial tumor with MN1 alteration”, with a calibrated score of 0.994 (Fig.3A). Copy number evaluation using data from the methylation array demonstrated alterations in chromosome 22 and X (Fig.3B). Subsequent gene fusion testing found a Ewing Sarcoma breakpoint region 1/EWS RNA binding protein 1 (EWSR1) - BEN Domain Containing 2 (BEND2) fusion between loci on chromosomes 22 and X (Fig.3C). The integrated diagnosis was malignant neoplasm consistent with astroblastoma with MN1 alteration.
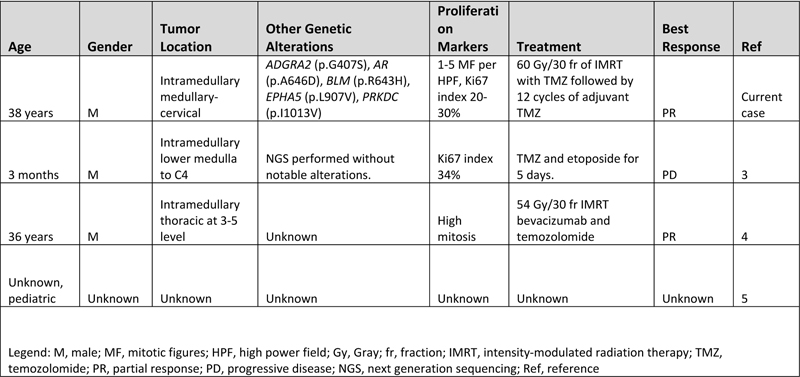
Figure 3. Methylation and copy number profiling of astroblastoma with an EWSR1-BEND2 fusion. (A) The lesion (blue box) was plotted on an X–Y coordinate graph (red dot to the lower left of the x–y intersection), where closer proximity to other dots indicates greater similarity of the index tumor’s genomic CpG methylation pattern to existing cases in the library. (B) Copy number profiling demonstrates the location for EWSR1-BEND2 fusion at chromosome 22 and X (blue arrow). (C) Fusion between exon 7 of EWSR1 and exon 5 of BEND2 genes with breakpoints at genomics positions chr22:29683123 and chrX:18234853 respectively. The red arrow represents the direction of the gene specific primary utilized by the Archer assay to enrich the amplicons for EWSR1 fusion events. Legend: MB,G3, medulloblastoma, subclass group 3; EPN, PF A, ependymoma, posterior fossa group A; EPN, PF B, ependymoma, posterior fossa group B; SUBEPN, PF, subependymoma, posterior fossa; MB, SHH CHL AD, medulloblastoma, subclass SHH A (children and adult); MB, SHH INF, medulloblastoma, subclass SHH B (infant); CONTR, CEBM, control tissue, cerebellar hemisphere; LIPN, cerebellar liponeurocytoma; CNS NB, FOXR2, CNS neuroblastoma with FOXR2 activation; SP-EPN-MYCN, MYCN amplified spinal cord ependymoma; EPN, YAP, ependymoma, YAP fusion; HGNET, MN1; high grade neuroepithelial tumor with MN1 alteration; GBM, G34, glioblastoma, IDH wildtype, H3.3 G34 mutant; GBM, MES, glioblastoma, IDH wildtype, subclass mesenchymal; GBM, MID, glioblastoma, IDH wildtype, subclass midline; GBM, MYCN, glioblastoma, IDH wildtype, subclass MYCN; GBM, RTK I, glioblastoma, IDH wildtype, subclass RTK I; GBM, RTK II, glioblastoma, IDH wildtype, subclass RTK II; GBM, RTK III, glioblastoma, IDH wildtype, subclass RTK III; HGNET, BCOR, CNS high grade neuroepithelial tumor with BCOR alteration; PTPR, A, papillary tumor of the pineal region group A; PTPR, B, papillary tumor of the pineal region group B; CONTR, PONS, control tissue, pons; CONTR, WM, control tissue, white matter; CONTR, HYPTHAL, control tissue, hypothalamus; CONTR, HEMI, control tissue, hemispheric cortex; CN, central neurocytoma; SUBEPN, SPINE, subependymoma, spinal; LGG, MYB, low grade glioma, MYB/MYBL1; LGG, DNT, low grade glioma, dysembryoplastic neuroepithelial tumor; LGG, GG, low grade glioma, ganglioglioma; IHG, infantile hemispheric glioma; LGG, PA/GG ST, low grade glioma, rosette forming glioneuronal tumor; LGG, RGNT, rosette forming glioneuronal tumor; CONTR, REACT, reactive tumor microenvironment; DLGNT, diffuse leptomeningeal glioneuronal tumor; ANA PA, anaplastic pilocytic astrocytoma; PXA, (anaplastic) pleomorphic xanthoastrocytoma; LYMPHO, lymphoma; DMG, K27, diffuse midline glioma H3 K27M mutant; LGG, PA PF, subclass posterior fossa pilocytic astrocytoma; LGG, PA MID, midline pilocytic astrocytoma; CONTR, INFLAM, control tissue, inflammatory tumor microenvironment; EPN, RELA, ependymoma, RELA fusion; ATRT, TYR, atypical teratoid/rhabdoid tumor, subclass TYR; CHGL, chordoid glioma of the third ventricle; LGG, SEGA, subependymal giant cell astrocytoma. Subsequent Clinical Course The patient was treated with fractionated radiation (5040 cGy in 28 fractions) with concurrent daily temozolomide (75 mg/m2) followed by adjuvant temozolomide (150-200 mg/m2, 5 days-on/23 days-off) with a partial response. At the time of writing, the patient has not progressed since initiation of treatment over one year ago. Clinically the patient was able to return to work full time with continued central neuropathic pain of the left face and right-side extremities and trunk managed with gabapentin. Discussion Astroblastoma has historically been a controversial entity since its introduction in the 1924 classification of CNS brain neoplasms by Cushing and Bailey.1 MN1-altered astroblastomas arise primarily in cerebral locations in pediatric patients and young adults.2 In addition to the presence of a EWSR1-BEND2 fusion, our case is unusual compared to other described MN1-altered astroblastoma in that the patient is near his fourth decade of life and with tumor located in the brainstem and upper cervical spine, rather than the cerebrum. Including the case presented, there are three other described cases of CNS tumors with a EWSR1-BEND2 fusion (Tab. 1).3–5 Aside from one case that did not provide additional information, these tumors occurred in males and were infratentorial, involving the spinal cord and sometimes the brainstem.3–5 Although non-specific, MRI of astroblastomas have been described to enhance, as well as appear well-demarcated, cystic, and lobulated, which is consistent with our case and similar to other cases of EWSR1-BEND2 fused astroblastoma tumors (Fig.1).3,4,6 Comparable to conventional MN1-altered astroblastomas and other cases of EWSR1-BEND2 fused tumors, our case showed perivascular growth and immunohistochemistry was positive for S100, OLIG2, EMA and GFAP.2–4 Ultrastructural observations in astroblastomas suggest a relationship to tanycytes, an ependymal cell subtype, but it is unclear if this applies also to MN1-altered and EWSR1-BEND2 fused astroblastomas.1,2 Molecular profiling has had a huge impact on the diagnosis of “astroblastic” tumors and has helped clarify subtypes of astroblastoma. In 2016, Sturm et al. identified a group of 41 tumors with a common methylation profile and MN1 fusions, designating them ‘CNS high-grade neuroepithelial tumor with MN1 alteration’ (CNS-HGNET-MN1).7 Notably, the majority of these contained astroblastic or ependymal perivascular pseudorosettes, although a number of other histopathological appearances were also represented. Subsequent studies of MN1-altered brain tumors have confirmed that many, but not all, have ependymal or astroblastic features.8 Based on these and other studies, the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy (cIMPACT-NOW) has proposed designating these tumors “astroblastoma, MN1-altered”.2 Early case series of astroblastoma reported prior to these newer molecular tools likely contained other tumor subtypes, as exemplified by a recent study in which molecular profiling of 14 adult tumors with histologic feature of astroblastoma found they were pleomorphic xanthoastrocytomas or high grade gliomas with alterations activating the mitogen-activated protein kinase pathway, and none had clear evidence of a MN1-BEND2 fusion, or clustered with that group on methylation profiling analysis.9 The MN1 alteration is usually a fusion between MN1 and BEND2.2 While half or more of microscopically defined astroblastomas harbor an MN1 alteration, a significant number do not.3,10 Our patient is unique due to the presence of a rare non-canonical EWSR1-BEND2 fusion between chromosomes 22 and X. Interestingly, early molecular analyses of astroblastomas identified deletions in chromosomes 22q and X.1,2 Including our case, there are at least four known cases of CNS tumor with EWSR1-BEND2 fusions in the literature (Table 1).3–5 One case was a 3-month -old male with tumor spanning the lower medulla to the C4 spinal cord level,3 with a second in a 36-year-old male patient with a thoracic spinal “ependymoma” with EWSR1-BEND2 fusion.4 A third case was found in a series of molecularly characterized pediatric tumors which did not provide clinical information.5 Remarkably, in at least two of these cases, as in ours, methylation clustering led to the tumor falling into the category of astroblastoma/HGNET with MN1 alteration.3,4 These reports highlight the possibility of astroblastic or ependymal tumors with a methylation profile consistent with “astroblastoma, MN1-altered” being driven by fusions in genes other than MN1, and also suggest that it may be BEND2, rather than MN1, which has a more critical functional role. Supporting this notion, there is one reported case of an neuro-epithelial tumor with an MN1-PATZ1 fusion found with RNA sequencing a supratentorial mass in a 1-year-old girl that suggest a similar pathogenic role as a chimeric protein.11 However, this case lacked astroblastomatous rosettes and did not have epigenetic similarities with CNS HGNET-MN1, but did have similarities to PATZ1-sarcomas.11 Table 1: Known Cases of Astroblastoma with an EWSR1-BEND2 fusion in the Literature
The grading and clinical behavior of astroblastomas is not well understood. As noted by the 6th cIMPACT-NOW update and other case series, a significant proportion of tumors in the methylation class Astroblastoma/CNS-HGNET-MN1 do not conform to astroblastoma histologically, and it is unclear if disparate histologic patterns have biologic relevance aside from high grade features.1,2 The 2016 WHO, completed before identification of the MN1-altered molecular group, does not assign grades, but observed that astroblastic neoplasms generally fall into two general categories: well-differentiated or anaplastic/malignant.1,12 Case series that predated molecular evaluation of astroblastomas likely included other tumor subtypes, but found that in histologically defined astroblastomas, an elevated proliferation rate is associated with worse outcomes.1,12 A case series of 14 neuroepithelial tumors with MN1 alterations, including three spinal cases, two of which were the oldest (14.6 and 36 years old) and only males in the series, describes heterogeneous treatments consisting of focal or cranial spinal radiation without chemotherapy.8 The event-free survival ranged considerably from 6 to 100 months, with some cases resulting in metastasis in the CNS, arguing that intermittent monitoring with completed neuroaxis imaging is warranted.8 A meta-analysis of 73 patients with MN1-altered neuroepithelial tumors (astroblastomas) found a 5- and 10-year progression-free survival of 38% and 0%, and 5- and 10-year overall survival of 89% and 55%, respectively.13 The natural history of astroblastoma with EWSR1-BEND2 fusion has not been well described in the literature, and it is unclear if the clinical behavior is significantly different from astroblastomas with a MN1 alteration. The case we present showed anaplastic microscopic features, as well as aggressive clinical behavior with rapid growth over two months - histologic features and clinical behavior was similar to other described cases of astroblastoma with a EWSR1-BEND2 fusion.3,4 Given the rarity of these tumors, there is no defined standard established treatment. Based on studies of non-molecularly characterized astroblastoma, resection remains the cornerstone of therapy to improve outcomes, as supported by an analysis of 116 patients that found that gross total resection improved outcome with a 5-year progression-free survival of 83% versus 55% in those with subtotal resection.1 Regarding radiation, a systemic review of 95 histologic astroblastoma patients did not show a survival benefit with radiation therapy; however, the series had a broad age range from 1 to over 61 years of age and did not compare survival differences in astroblastoma with and without anaplastic features.14 Review of the literature found that a 20-year-old woman with a spinal cord MN1-altered astroblastoma, and a 36-year-old man with a spinal astroblastoma with EWSR1-BEND2 both had a reduction in tumor size after radiation, temozolomide, and bevacizumab.4,15 In another case, a 3-month old boy with EWSR1-BEND2 astroblastoma had progressive disease after five days of temozolomide and etoposide.3 Given the significant growth of the presented patient's tumor in two months (Fig.1) and dramatically elevated Ki67 proliferation index, he was treated with concurrent radiation with temozolomide followed by adjuvant temozolomide based on the “Stupp” protocol and this appears to be a viable option, as he has had a partial response control of his tumor over one year later at the time of writing.16 This case exemplifies the use of advanced molecular testing, including the evaluation of chromosome fusions and methylation profiling, to accurately diagnose rare neoplasms of the CNS. Through these technologies, improved diagnosis accuracy provides clinically impactful insights that enhance the understanding of tumor biology. Funding This work was supported by the National Institutes of Health T32 research training grant. Authors’ Contributions M.A.S case report concept, design, and critical revision of content. Z.A., K.D.A, M.Q., M.K.R., F.J.R., C.G.E and L.J.L. provided critical revision of content. Acknowledgments The authors thank Rust Turakulov at the NIH for help with formatting the methylation plot for publication. The authors also thank the patient and his caregivers. References 1. Brat, D. J., Hirose, Y., Cohen, K. J., Feuerstein, B. G. & Burger, P. C. Astroblastoma: Clinicopathologic Features and Chromosomal Abnormalities Defined by Comparative Genomic Hybridization. Brain Pathology 10, 342–352 (2006). 2. Louis, D. N. et al. cIMPACT-NOW update 6: new entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol bpa.12832 (2020) doi:10.1111/bpa.12832. 3. Yamasaki, K. et al. Spinal cord astroblastoma with an EWSR1-BEND2 fusion classified as a high-grade neuroepithelial tumour with MN1 alteration. Neuropathol Appl Neurobiol 46, 190–193 (2020). 4. Tsutsui, T. et al. PATH-23. ADULT SPINAL CORD ASTROBLASTOMA WITH EWSR1-BEND2 FUSION. Neuro-Oncology 22, iii429–iii429 (2020). 5. Ramkissoon, S. H. et al. Clinical targeted exome-based sequencing in combination with genome-wide copy number profiling: precision medicine analysis of 203 pediatric brain tumors. Neuro Oncol 19, 986–996 (2017). 6. Sener, R. N. Astroblastoma: diffusion MRI, and proton MR spectroscopy. Comput Med Imaging Graph 26, 187–191 (2002). 7. Sturm, D. et al. New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164, 1060–1072 (2016). 8. Baroni, L. V. et al. Treatment response of CNS high-grade neuroepithelial tumors with MN1 alteration. Pediatr Blood Cancer 67, (2020). 9. Boisseau, W. et al. Molecular Profiling Reclassifies Adult Astroblastoma into Known and Clinically Distinct Tumor Entities with Frequent Mitogen-Activated Protein Kinase Pathway Alterations. Oncologist 24, 1584–1592 (2019). 10. Mhatre, R. et al. MN1 rearrangement in astroblastoma: study of eight cases and review of literature. Brain Tumor Pathol 36, 112–120 (2019). 11. Burel-Vandenbos, F. et al. A polyphenotypic malignant paediatric brain tumour presenting a MN1-PATZ1 fusion, no epigenetic similarities with CNS High-Grade Neuroepithelial Tumour with MN1 Alteration (CNS HGNET-MN1) and related to PATZ1 -fused sarcomas. Neuropathol Appl Neurobiol 46, 506–509 (2020). 12. Bonnin, J. M. & Rubinstein, L. J. Astroblastomas: a pathological study of 23 tumors, with a postoperative follow-up in 13 patients. Neurosurgery 25, 6–13 (1989). 13. Chen, W. et al. Central nervous system neuroepithelial tumors with MN1-alteration: an individual patient data meta-analysis of 73 cases. Brain Tumor Pathol 37, 145–153 (2020). 14. Sughrue, M. E. et al. Clinical features and post-surgical outcome of patients with astroblastoma. J Clin Neurosci 18, 750–754 (2011). 15. Yamada, S. M. et al. Primary spinal cord astroblastoma: case report. Journal of Neurosurgery: Spine 28, 642–646 (2018).
Copyright: © 2021 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |