|
|
|
Free Neuropathology 1:32 (2020) |
|
Review |
|
Neuropathology of the Alzheimer’s continuum: an update |
|
Kurt A. Jellinger |
|
Institute of Clinical Neurobiology, Vienna, Austria |
|
Address for correspondence: |
|
Submitted: 06 October 2020 Accepted: 07 November 2020 Copyedited by: Nicole Schwab Published: 11 November 2020 |
|
Keywords: Alzheimer’s disease, β-Amyloid, Tau pathology, Oligomers, Amyloid angiopathy, Alzheimer subtypes, Regional vulnerabiliy, Co-pathologies |
|
Abstract Alzheimer’s disease (AD), the most common form of dementia worldwide, is a mixed proteinopathy (amyloid and tau). Originally defined as a clinicopathological entity, it is a heterogenous, multifactorial disorder, currently referred to as the Alzheimer’s continuum. Its cardinal pathological features are extracellular β-amyloid (amyloid plaques) and intraneuronal tau aggregates forming neurofibrillary tangles, which are accompanied by vascular amyloid deposits (cerebral amyloid angiopathy), synapse and neuronal loss, as well as neuroinflammation and reactive astrogliosis. In addition to “typical” AD, various subtypes with characteristic regional patterns of tau pathology have been described that show distinct clinical features, biomarker levels, and patterns of key network destructions responsible for cognitive decline. AD is frequently associated with other age-related changes including Lewy and TDP-43 pathologies, hippocampal sclerosis, argyrophilic grain disease, cerebrovascular lesions, and others. These additional pathologies influence the clinical picture of AD, may accelerate disease progression, and can cause a number of challenges in our understanding of the disease including the threshold of each individual pathology to cause dementia and the possibility of underlying common etiologies. This article provides an up-to-date overview of AD neuropathology, its heterogeneity, and additional pathologies in order to explain the difficulties in the diagnosis and the failure of clinical trials in AD patients. Abbreviations AD – Alzheimer’s disease, ADNC - Alzheimer’s disease neuropathological changes, AP - amyloid plaque, APP - amyloid precursor protein, Aβ - β-amyloid peptide, AβO - Aβ oligomer, CAA - cerebral amyloid angiopathy, CBS - corticobasal syndrome, CERAD - Consortium to Establish a Registry for Alzheimer Disease, CSF - cerebrospinal fluid, CVD - cerebrovascular disease, DLB - dementia with Lewy bodies, EOAD - early-onset AD, FTLD - frontotemporal lobar degeneration, FTLD-TDP - frontotemporal lobar degeneration with TDP-43, GVD - granulovacuolar degeneration, HcSp-AD - hippocampal sparing AD, hp-tau - hyperphosphorylated tau protein, LATE - limbic-predominant age-related TDP-43 encephalopathy, LATE-NC - limbic-predominant age-related TDP-43 encephalopathy neuropathological change, LC - locus ceruleus, LOAD - late-onset AD, LP-AD - limbic-predominant AD, LPPA - logopenic primary progressive aphasia, MA-AD - minimal-atrophy AD, MCI - mild cognitive impairment, MTL - medial temporal lobe, NFT - neurofibrillary tangle, NIA-AA - National Institute of Aging/Alzheimer's Association, NP - neuritic plaque, NT - neuropil thread, PART - primary age-related taupathy, PCA - posterior cortical atrophy, PHF - paired helical filament, SF - straight filament, TDP-43 - 43-kDa TAR DNA binding protein 43. 1. Introduction Alzheimer’s disease (AD) is the most common form of dementia, currently affecting around 50 million people worldwide. It accounts for 60-70% of dementia cases in clinical and autopsy series, but it is often associated with other confounding pathologies in the elderly. Its incidence increases from 2/1.000 at age 65-74 years to 37/1.000 at age 85+ [1], and doubles every five years after age 65, with peaks in the tenth decade and slight decrease afterwards [2, 3]. The point prevalence of AD among individuals aged 60+ is 40.2/1,000 persons, the pooled annual period prevalence is 30.4/1,000, and the incidence rate is 15.8/1,000 person-years [4]. With the disproportional increase of the elderly population, the prevalence of AD will approach around 132 million worldwide and up to 16 million cases in the USA by 2050 [5, 6], AD has become a tremendous public health and socio-economic challenge of the 21st century [5]. As available treatments only target symptoms and neither slow nor reverse the progression of the disease, the development of disease-modifying therapeutic procedures is urgent [7]. AD was originally defined as a clinicopathological entity, characterized by progressive memory deficit, involvement of multiple cognitive domains, and a defining pathological substrate with deposition of amyloid-β peptide (Aβ) in extracellular plaques and cerebral vasculature (cerebral amyloid angiopathy/CAA), neuritic plaques defined by the presence of microtubule-associated hyperphosphorylated tau protein (hp-tau), intraneuronal aggregations of hp-tau manifesting as neurofibrillary tangles (NFTs) in the cell soma, and neuropil threads (NTs), which occur mainly in dendritic compartments and, to a lesser degree, in the axonal domain. These changes are accompanied by early synaptic loss [8], activated microglia [9], mitochondrial dysfunction causing energy loss [10], neuroinflammation [11], neurovascular dysfunction [12], disruption of the blood-brain barrier [13], neuronal loss and reactive astrogliosis [14]. AD, a mixed proteinopathy (amyloid, tau, TDP-43, and others), is a heterogenous disorder currently referred to as the Alzheimer’s continuum [15] with several pathobiological subtypes and various co-pathologies [16]. The final definite diagnosis of AD rests with post-mortem neuropathology despite the advent of more sensitive neuroimaging and the use of reliable biomarkers [17]. Even though the classical morphological features of AD have been known for many years, the recently used more sensitive immunohistochemistry techniques for Aβ and hp-tau have replaced silver-staining techniques and have not only forwarded the diagnosis of AD but allowed a more scientific evaluation of the disease's pathology. For the neuropathological diagnosis of AD, the updated National Institute on Aging/Alzheimer's Association (NIA/AA) 'ABC' criteria are used [17]. The morphological changes involving brain regions and neuronal cell types following a stereotypical pattern [18] result from selective cellular and regional vulnerability to pathogenic factors and their progression through functionally integrated regions of the brain [19-23] as well as functional networks that result in progression of AD [24, 25]. However, AD is a heterogenous continuum with a variety of clinically and morphologically defined subtypes, currently referred to as Alzheimer’s clinical syndrome [15], which presents major challenges for both diagnosis of AD, monitoring and targeting of disease progression [26]. The new definition of AD as a biologically defined spectrum, using the NIA/AA framework [15], enables recognition and diagnosis of the various subtypes of AD [16]. Research consensus guidelines have been proposed for the intra vitam biologically-based categorization termed 'ATN', which uses combinations of in vivo biomarkers for Aβ deposition (A), tau pathology (T), and neurodegeneration (N). They use cerebrospinal fluid (CSF) or plasma biomarkers, PET, and functional and structural MRI. The biomarker profiles and categories of the Alzheimer’s spectrum referring to AD neuropathological changes (ADNC) have been summarized recently [27]. 2. Pathology of Alzheimer’s disease 2.1. Macroscopic features The AD brain often has decreased weight and at least moderate cortical atrophy most marked in the medial temporal lobes (MTLs) with relative sparing the primary motor, somatosensory and visual cortices and enlargement of the lateral ventricles (ex vacuo hydrocephalus). Brain atrophy often involves posterior cortical areas, most notable in precuneus and posterior cingulate gyrus in the preclinical stage of AD [28]. However, none of the macroscopic features are specific to AD, and healthy elderly people often show moderate cortical atrophy especially affecting the frontal lobes, with volume loss of the white matter [29]. Medial temporal atrophy affecting amygdala and hippocampus with enlarged temporal horn is typical of AD (Fig. 1). However, this is also seen in other age-related disorders such as hippocampal sclerosis [30].
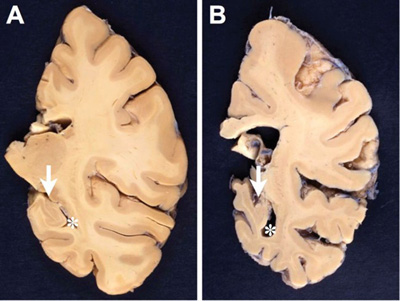
Figure 1. Comparison between formalin-fixed brain slices of the left hemispheres (level of posterior hippocampus) of an aged nondemented individual (A) and an AD patient (B). Note the marked atrophy (thinning of the gyri and deepening of the sulci) in B, in particular hippocampal atrophy (arrow in B) with widening of the inferior horn of the second ventricle (asterisk in B). Photographs by courtesy of Simon Fraser and Arthur Oakley. 2.2. Microscopic features The definite diagnosis of AD requires microscopic examination of multiple brain regions with semiquantitative assessment of the density of lesions and their topographical distribution. Extracellular amyloid plaques (APs) and intracellular NFTs that are essential for the neuropathological diagnosis, are associated with tau-positive NTs, dystrophic neurites and neuritic plaques (NPs), CAA, reactive astrocytes and activated microglia, and neuroinflammation are present. These lesions result in loss of synapses and neurons in vulnerable regions leading to brain atrophy and the characteristic clinical picture of the disease. Hirano bodies, granulovacuolar degeneration (GVD), TDP-43 deposits, and other lesions may also be present [31, 32]. 2.3. Amyloid deposits APs are formed by the abnormal extracellular nonvascular accumulation and deposition of Aβ peptides of varying length including those with 40 or 42 amino acids (Aβ-40 and Aβ-42), resulting from the sequential cleavage of the amyloid precursor protein (APP) by the enzymes β- and γ-secretases [33]. APP, from which Aβ is cleaved by endoproteolytic processing, is a large single transmembrane protein, encoded by the APP gene on chromosome 21 [34, 35]. Proteolytic cleavage of APP develops mainly via two exclusive pathways, the amyloidogenic and the non-amyloidogenic pathway, but other alternative pathways (η-secretases, δ-secretase, etc.) have been described for the physiological processing of APP [36]. The initial cut at the β-site of APP is due to the β-secretase activity enzyme BACE1, a transmembrane enzyme with aspartyl protease activity. Clearance by β-secretase yields a slightly shorter soluble fragment (sAPPβ) and a correspondingly longer C-terminal fragment (CTFβ) or C99 [37]. APP undergoes constitutive shedding by a protease activity called α-secretase, which appears to be a metalloprotease of the ADAM family. TACE (ADAM17) is one of the α-secretase, but ADAM10 is more important for α-secretase activity and sAPPα production. ADAM10 is the physiologically relevant constitutive of α-secretase in primary neurons [38], as has been demonstrated in vitro and in vivo [39]. Cleavage of APP by α-secretase releases the soluble ectodomain of APP, called sAPPα, and a membrane-tethered intracellular C-terminal fragment, termed CTFα of C83. The amyloidogenic (or β) cleavage of APP is in direct competition with an alternative non-amyloidogenic pathway (cleavage by the α-secretase within the Aβ sequence) which precludes the formation of amyloidogenic peptides and leads to soluble sAPPα, and has neuroprotective properties preventing Aβ production [40]. However, aberrant sAPPα production may tilt the cells toward unregulated growth, but the underlying mechanisms are still unknown [41]. Lastly, γ-secretase, a high molecular weight complex that consists of presenilin (PS1, PS2), an aspartyl membrane protease, Aph-1, nicastrin and presenilin enhancer (PEN2), cleaves APP terminal fragments (CTFs) such as C83 and C99, releasing 3 or 4 amino acid peptides from the transmembrane fragment of APP. Notably, γ-secretase is active on APP only following the antecedent α- or β-secretase. The products of γ-secretase cleavage of C83 are a 3-kDA peptide, termed p3 and an APP intracellular domain (AICD), while γ-secretase cleavage of C99 yields the infamous Aβ peptide and an identical AICD fragment. Besides cleavage by α-, β-, and γ-secretase, other N-terminal fragments (NTFs) of APP have been identified that are generated by unknown proteases [42, 43]. Mounting evidence suggests that astrocytes that have increased levels of APP, β-secretase (BACE1), and γ-secretase play an additional role in AD by secreting significant amounts of Aβ and contributing to overall Aβ burden in the brain [44]. BACE1 inhibition more effectively suppresses the initial process of plaque formation, rather than the subsequent phase of plaque growth, which has implications for therapeutic efficiency for the treatment of AD [45]. AD is driven by intraneuronally retained Aβ produced by the AD-specific βAPP-independent pathway [46]. Neuronal Aβ-42 is enriched in small vesicles at the presynaptic side of synapses [47]. Aβ deposits contain a mixture of various isoforms. The most common are Aβ-40 (under physiologic conditions around 90%), Aβ-38 and Aβ-42 (less than 10%). Aβ-40 is produced within the trans-Golgi network (TGN) whilst Aβ-42 can be made in either the TGN or the endoplasmic reticulum [48]. The specific production of Aβ-42 in the endoplasmic reticulum of neurons links this compartment with the generation of Aβ and explains why primarily endoplasmic reticulum localized proteins such as presenilin could induce AD [49]. Increased production of Aβ-42 at the expense of Aβ-40 is a common feature in both familial and sporadic AD [50]. The latter is believed to be more toxic than Aβ-40 because of its tendency to aggregate and to form fibrils [51]. The phosphorylation of APP by extracellular-regulated kinase (ERK) and protein kinase C (PKC), in the proteolytic processing of APP has been demonstrated to be critically modulating the generation of Aβ [52]. The C-terminal APP fragments (APP intracellular domain) are generated by γ-secretase cleavage [53]. γ-Secretase was shown to cleave near the cytoplasmic membrane boundary of APP, called ε-site cleavage, as well as in the middle of the membrane domain, called γ-site cleavage, indicating that γ- and ε-site cleavage are regulated independently [54]. Ubiquilin-1 has been shown to modulate γ-secretase-mediated ε-site cleavage and thus may play a role in regulating γ-secretase cleavage of APP and other proteins [55]. Further cleavage of APP intracellular domain (AICD) fragments by caspase or caspase-like proteases results in additional fragments which, however, does not seem to require antecedent proteolysis of APP [41].
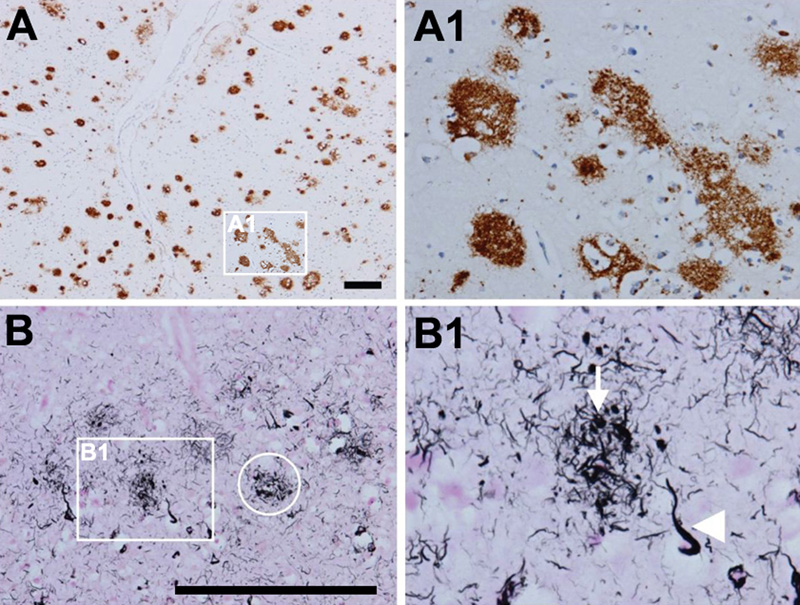
Figure 2. Amyloid and neuritic plaques. A; A1. Multiple diffuse amyloid plaques in the neocortex (antibody 4G8). B, B1. Neuritic plaques that contain Aβ and tau in distended processes (i.e. dystrophic neurites). Gallyas silver stain visualizes both aggregated Aβ and tau and is therefore ideal to detect neuritic plaques (ring in B, neuritic plaque; arrow in B1, dystrophic neurite; arrowhead in B1, neurofibrillary tangle). Scale bars: 200 μm. From [71]. Truncated Aβ fragments are deposited in APs due to axonal linkage and release of APP [56]. Chemical imaging of evolving AP pathology in a transgenic mouse model for AD suggested initial plaque formation to be seeded by Aβ-42, followed by plaque maturation upon deposition of Aβ-40 as well as deposition of others [57]. Due to its higher rate of fibrillization and insolubility, Aβ-42 is its major component in addition to other Aβ peptides [58]. A recent report demonstrated the role of HIF-1alpha/lncRNA BACE1-AS axis in the transactivator of transcription (Tat)-mediated induction of astrocytic amyloidosis [59]. Advanced biophysical examination of Aβ derived from AD brain tissue showed polymorphic structures [60]. The terminology of Aβ plaques is confusing, since a myriad of non-vascular Aβ deposits have been described, but five major types can be distinguished: (a) primitive or immature plaques are spherical deposits of predominantly Aβ-42 in the neuropil without a dense core and neurites; (b) diffuse plaques, usually large (50µm to several hundred µm), slightly immunoreactive and ill-limited, contain loose amyloid bundles in the neuropil without degenerating neurites and accompanying microglia (Fig. 2A); (c) stellate deposits probably related to astrocytes [61]; (d) focal deposits with dense and spherical accumulations of Aβ-42, surrounded by a neuritic corona containing dystrophic tau-positive neurites and astrocytic components, constituting the “cored”, “classical” or “neuritic” plaques (NPs) (Fig. 2B, 2B1); and finally (e) compact or burnt-out plaques with a dense core of Aβ-40, absent or tau-negative, ubiquitin-positive neurites. NPs have compact dense amyloid cores composed of more fibrillated forms of Aβ (Fig. 3). They contain tau-positive dystrophic neurites and are accompanied by synaptic loss, activated microglia and reactive astrocytes [62, 63]. There are differences in the composition of the aggregates, for example, the Aβ in NPs has a more varied composition with the presence of Aβ 40, 42, 43, N-terminus truncated Aβ and other post-transitionally modified forms [64, 65]. Tau-positive NPs begin early in AD, but major tau deposition follows the Aβ deposition and the clustering of activated microglia [66]. Recent studies unequivocally demonstrated that plaque-associated myeloid cells are derived exclusively from resident microglia [67]. In AD, microglia can eliminate APs through phagocytosis with APOE lipoprotein at an early stage of disease progression [68]. Scanning transmission electron microscopy (STEM) showed three types of fibrillary network structures: amorphous network, fibril bundles, and amyloid stars [69]. Although diffuse non-neuritic plaques are generally present before NPs, whether an individual diffuse plaque can actually transfer into an NP or whether these two types develop differently, is not clear at present. A recently described type called the coarse-grained plaque, a relatively large deposit (diameter about 80 µm) characterized by multiple cores and Aβ-devoid pores, is prominent in the neocortex and associated with homozygous APOEε4 status and CAA. This divergent AP type is similar to CAA, predominantly composed of Aβ-40, and has been observed particularly in early-onset AD (EOAD) [70].
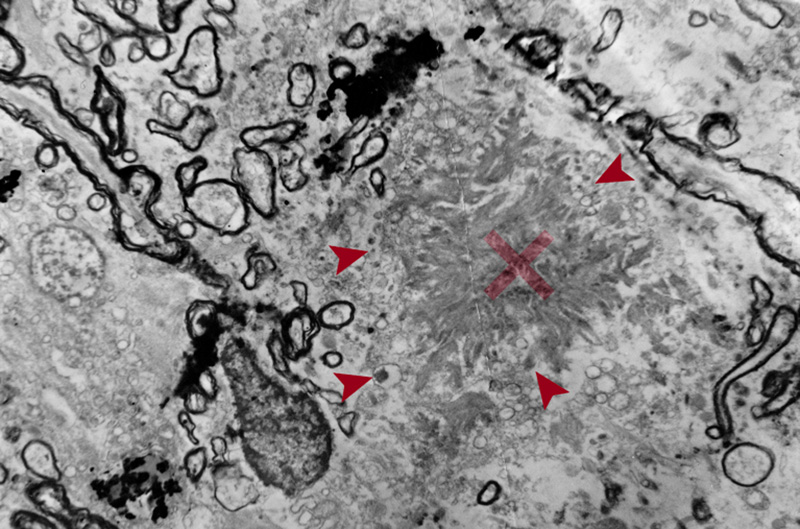
Figure 3. EM image of amyloid core of a neuritic plaque. Radiating bands of amyloid fibrils comprise the core (x). Note the adjacent abnormal fibrils filled with dense bodies (arrows) and surrounding damaged myelin sheaths (x 4000). “Burnt out” plaques are composed of dense cores lacking neuritic components, while the astrocytic processes penetrating the plaque core may represent a regressive stage (“remnant plaques”) [72]. “Cotton wool plaques” are non-compact deposits, made of Aβ-42 with sparse glial components and variable neurites but not surrounded by a neuritic corona. They can be detected with H&E staining [61]. Aβ and tau each begin to aggregate in separate neuroanatomical locations and meet in the cerebral cortex in the NP. This “collision” of both proteins mediated by microglia has devastating consequences in terms of neuronal loss, promoting neurodegeneration and the consequent development of cognitive decline, but this is still under investigation [73]. 2.4. Distribution of amyloid deposits APs in AD brain show a typical distribution with brain areas that are connected via the “default network” typically affected early. In animal models some demonstration of “propagation” along neuronal systems has been observed [74, 75], suggesting some axonal transport of seeds that lead to extracellular deposits. Most Aβ deposits are located in the gray matter, while some diffuse or lake-like deposits may be seen in the subpial white matter. Cortical soluble Aβ protein is a neurotoxic agent [76, 77], and Aβ oligomers (AβOs) may trigger the early phase of the Aβ seeding process, while depletion of AβOs delays the aggregation process leading to a transient reduction of seed-induced Aβ deposits [78]. The topography of Aβ deposits depends on the stage of the disease, which leaded to several staging schemes. Three stages were distinguished: Stage A with amyloid deposits in the basal portions of the frontal, temporal and occipital cortex; in stage B all isocortex is involved, with primary cortices spared and the hippocampus only mildly affected; while stage C shows deposits in the whole isocortex including sensory and motor core fields [18]. Others proposed five amyloid “phases” using sensitive silver staining or Aβ antibodies: stage 1 or isocortical, stage 2 with additional involvement of hippocampus and entorhinal cortex, stage 3 plus striatum and diencephalic nuclei, stage 4 several brainstem nuclei and medulla oblongata, and stage 5 presenting amyloid deposits in the pons and molecular layer of the cerebellum [79, 80]. These can be reduced to three stages: 1 - isocortical, 2 - allocortical or limbic, and 3 - subcortical. Usually involved is the total isocortex, layers II-V more than layers I and VI [18]. In advanced cases band-like diffuse Aβ deposits are also seen in the subpial surface of the cortex or in the white matter close to layer VI [71]. Amyloid PET-based staging of Aβ pathology in vivo confirmed its progression in AD [81], and revealed higher plaque counts in entorhinal and occipital regions of typical AD, while other phenotypes showed more severe Aβ deposition in frontal and parietal cortices [82]. Post-mortem analysis of (18)Fflutemetamol and (11)CPiB PET signal showed that it is influenced by both diffuse plaques and cored plaques and, therefore, is likely a function of plaque size and density of Aβ fibrils in plaques. Brain regions with large volumes of diffuse plaques could yield PET retention levels comparable with lower volume/frequency of cored plaques [83]. 2.5. Cerebral amyloid angiopathy Aβ peptides also involve the vessel walls, as with CAA, with the more soluble Aβ-40 as the major constituent. 85-90% of confirmed AD cases have some degree of CAA [84]. It mainly accumulates in the interstitium between the smooth cells of the tunica media. Small arteries, arterioles and even capillaries in the cerebral cortex and leptomeningeal vessels are affected [85]. Stage 1: vessels are affected in the isocortex, stage 2: involvement of allocortex, and stage 3: basal ganglia, thalamus, pons and medulla oblongata [86]. Others distinguished four patterns [87]: Type 1: APs with or without CAA in the leptomeninges alone; type 2: CAA in both leptomeningeal and deeper penetrating arteries (Fig 1A); type 3: CAA affects both precapillaries and arterioles; type 4 shows Aβ deposition in and around blood vessels. Genetically, type 3 (capillary subtype) is more strongly associated with the APOEε4 allele [87, 88]. Two other types were distinguished: Type 1 affecting capillaries, arterioles and small arteries is associated with APOEε4, whereas type 2 not involving capillaries is more likely associated with APOEε2, its most frequent form [89]. Both severe CAA and AD are associated with APOEε4-positive patients [88]. A more recent staging system is based on the severity of CAA in a single vessel: grade 0: absence of staining, grade 1: a congophilic ring around the otherwise normal-appearing vessel, grade 2: complete replacement of the tunica media by congophilic material, grade 3: involving >50% of vessel circumference, giving a “double-barrel” appearance, and grade 4 or fibrinoid necrosis of the vessel wall with additional amyloid deposits in the surrounding neuropil (“dyshoric changes”) [88]. The parietal and occipital cortices are more vulnerable than the frontal and temporal lobe, and the leptomeningeal vessels more than the parenchymal ones [84]. Aβ deposition shrinks the cerebral blood vessels by about 8% and reduces the energy supply resulting from decrease of blood flow [90]. CAA can cause small infarcts in the cerebral cortex, while severe CAA may lead to lobar hemorrhages in the frontal and occipital lobes and to diffuse white matter lesions (Fig. 4) [91]. Brain hemorrhage does not appear to be directly linked to amyloid burden in patients with CAA-related intracerebral hemorrhage, because amyloid burden was similarly distributed across the brain hemispheres and no interhemispheric difference was observed for Aβ burden nor for MRI markers of small vessel disease [92]. CAA and deep perforating arteriopathy are similar and interact with blood-brain barrier breakdown, endothelial damage, and impaired perivascular Aβ drainage. Both may cause ischemic lesions and intracerebral hemorrhages [93]. Chronic treatment of a mouse model of AD with fungicides produced Aβ fibril formation and impairment of Aβ clearance through neprylisin, suggesting that fungicide residues could be a risk factor for AD via CAA [94]. Although several pathogenic mechanisms, including the disbalance between production and clearance of Aβ creating a self-reinforcing cycle of increased vascular Aβ and further CAA and AD progression, have been shown, they do not explain completely the disease pathogenesis [95]. The intersection between CAA and AD points to a crucial role for improving vascular function in the treatment of AD [96].
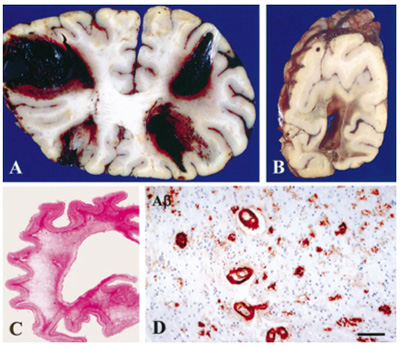
Figure 4. Multiple large hemorrhages in both frontal lobes (A) and occipital lobe (B). Diffuse white matter destruction (C). CAA in many vessels in the cerebral white matter; scale bar 70 µm (D). From [97]. 2.6. Tau pathology Tau protein is encoded by the MAPT (microtubule-associated protein tau) gene on chromosome 17 [98], which generates a total of 6 isoforms through alternative splicing of exons 2, 3 and 10 in the CNS [99]. Tau protein, the main constituent of NFTs, is involved in the stabilization of neurotubules that leads to the appropriate function of the neuron. Its microtubule-binding regions are made of 3 or 4 repeats (3R or 4R tau), their second repeat (exon 10) being spliced in some isoforms. Combined phosphorylation of Ser202, Thr205, and Ser208 forms a unique post-translational modification configuration that promotes tau aggregation, accelerating the formation of tau filaments and eventually resulting in NFT formation. Tau adopts different stable conformations, consistent with the notion of 'strains' as may be seen with the concept of phenotypic diversity or with different environmental stimuli [100, 101]. Truncation of tau by caspases-3 or -4 is an early event in the development of NFTs [102]. The molecular mechanisms leading to the accumulation of tau are characterized by numerous translational modifications that change its conformation and structural state. Recent studies indicate that the dysregulation and dislocation of splicing factor proline and glutamine rich (SFPQ), the subsequent DNA anomalies and aberrant dynamics of TIA-1-positive stress granules in association with pathological tau may represent a critical pathway which contributes to the rapid progression of AD [103]. Abberant phosphorylation and truncation make tau protein into a pathological entity; paired helical filaments (PHF), the major structural constituents of NFTs, exhibit a greater degree of phosphorylation than normal tau [104].Tau monomers can aggregate to form oligomers and higher-order fibrils. Whilst Aβ can largely self assemble, tau phosphorylation is believed to be important for its aggregation [105]. Phosphorylation of Ser208 likely occurs at different disease stages from phosphorylation of Ser202 and Thr205. hp-Tau accumulation causes synaptic impairment, neuronal dysfunction, and formation of NFTs. Tau with site-specific posttranslational modification/soluble hp-tau species impact mitochondria and facilitate neurodegeneration [106]. Recent studies support the hypothesis that tau phosphorylation at Ser208 strongly contributes to unique types of tau aggregates, and may be a reliable marker for the presence of mature NFTs [107]. In AD, tau protein usually accumulates in the somato-dendritic and, to a lesser degree, in the axonal domains of the neuron. NFTs and pretangles are due to accumulation in the soma; NTs occur in dendrites, and the neuritic corona of core plaques is constituated by axonal processes filled by tau proteins (Fig. 2). As major constituents of NFTs and NTs, they are hyperphosphorylated and aberrantly misfolded, have lost their microtubule stabilizing functions, and contribute to axonal transport deficits [105]. PHFs in AD contain all 6 isoforms of tau protein including those with 3 and 4 repeats (3R- and 4R-tau) in the microtubule binding domain, forming the core of PHF [108]. The tau isoforms show a chronological shift: initially, early pretangles are positive only for 4R, gradually 3R is involved in mature tangles, and finally 4R is replaced by 3R in ghost tangles [109]. Ultrastructurally, NFTs appear as PHFs, i.e., fibrils of ca. 28 nm in diameter that form pairs with a helical tridimensional conformation and a regular periodicity of 65-80 nm [110] or as helical or twisted ribbons [111]. Straight filaments (SFs) show a longer crossover distance and modulations in width from 10 to 15 nm. Both lesions are different from those seen in other tauopathies [112]. PHFs and SFs differ in their inter-protofilament packing, and are ultrastructurally polymorph [113]. Visible with cryo-EM, PHFs and SFs are made of two C-shaped protofilaments with a combined cross-β-β-helix structure, without variations in the filamentous structures between sporadic and inherited AD [114]. NTs have an ultrastructure and immunohistochemistry similar to NFTs. Why, despite its axonal origin, PHF tau accumulates primarily in the neuronal cell body and dendrites, is unknown. It shows in three stages: (a) Pre-NFTs composed of diffuse, or punctuate tau staining occur within the cytoplasm of otherwise normal-looking neurons with well-preserved neurites; or (b) mature intraneuronal NFTs consist of cytoplasmic filamentous aggregates of tau displacing the nucleus toward the periphery of the soma and extending to the proximal segment of the axon. They appear as “flame-shaped tangles” in pyramidal neurons of the hippocampus (Fig. 5) and layer V of association cortices and as “globose tangles” in subcortical nuclei; (c) extraneuronal “ghost” NFTs in dead neurons, showing loss of their nucleus and of stainable cytoplasm [115]. Total loss of functional microglia in advanced late-onset AD (LOAD) promotes widespread intraneuronal neurofibrillary degeneration leading to brain failure [116]. Neuronal tau pathology has been linked to neuronal death and cognitive decline in AD [117], while others suggested that neuronal cell loss is associated with dementia and not the presence of plaques and tangles [118]. It is generally thought that NFTs impede neuronal functioning, but recent data indicate that they can be found in functionally intact neurons integrated in cortical circuits [119-121]. How hp-tau specifically mediates its toxic effects is still unknown, but oligomeric tau species, analogues to AβOs, are potential toxic species besides NFT tau. Recent proteomic studies have identified specific proteins that interact with hp-tau, showing novel potential pathogenic mechanisms that are relevant in AD and providing insight into how hp-tau mediates its toxicity in AD [122].
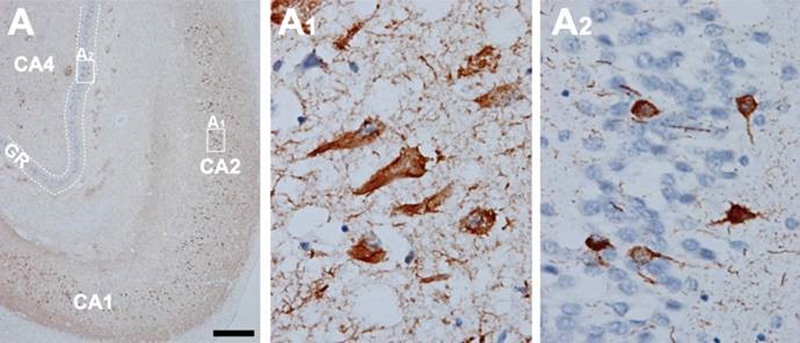
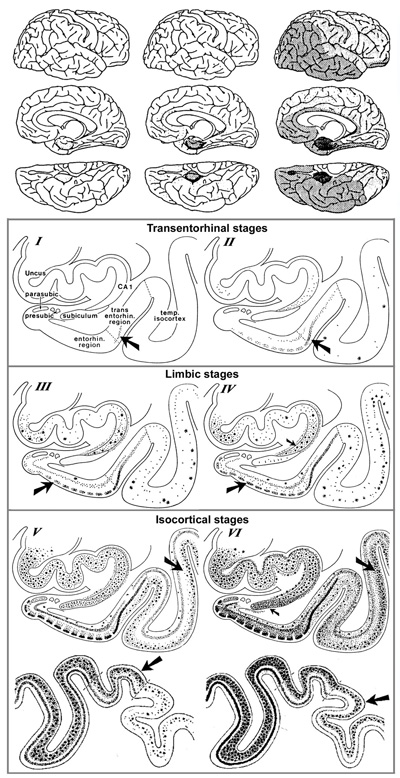
Figure 5. In AD, high amounts of neurofibrillary tangles and neuropil threads are seen in the hippocampus (A). CA1, CA2, and CA4 hippocampal cornu ammonis (Ammon’s horn) sectors 1, 2, and 3, respectively; GR, granule cell layer of the dentate gyrus. Immunohistochemistry with antibody AT8. Scale bar: 50 μm. From [71]. 2.7. Topography and spreading pattern of tau The extent of tau pathology (NFTs and NTs) follows a predictable spatiotemporal progression through functionally integrated brain regions [18], which had been interpreted as a cell-to-cell spreading through prion-like propagation [123-127] or a transneuronal spread through functional networks, associated with a trigger, possibly Aβ and/or neuronal network activity that could lead to progression of NFT pathology [128]. Since tau is expressed predominantly in neurons rather than glial cells, the detection of tau aggregates in astrocytes and oligodendroglia has given support to the concept that the release of misfolded tau from neurons (or oligodendroglia) may result in uptake into other cells [129]. Microglia could potentially play a role in spreading of tau pathology [130]. Transcellular progression of tau seeds has been observed in early Braak stage in regions predicted to be free of hp-tau [131]. According to the original staging [18], the first NFTs consistently occur in the transentorhinal (perirhinal) region (stage I) along with the entorhinal cortex, followed by the CA1 region of the hippocampua (stage II), indicating a preclinical phase of AD which can last up to 20 years. Limbic structures, such as the subiculum of the hippocampal formation are affected next (stage III), followed by the amygdala, thalamus, and claustrum (stage IV). Stages III and IV are often correlated clinically with mild cognitive impairment (MCI). In stage V, NFTs spread to isocortical areas with the association areas being affected prior and more severely, followed in stage VI by the primary sensory, motor and visual areas, which is usually associated with overt dementia (Fig. 6). This NFT staging has been widely accepted in routine pathology and appears well correlated with the clinical status, at least in the amnestic AD. Imaging in vivo tau pathology with tau-specific PET tracers identified NFT pathology reflecting Braak stages IV or higher. It rendered it possible to study the temporal progression of tau pathology in vivo, and, therefore, can be used as a reliable biomarker of tau pathology [132-137]. There is an inverse correlation between the accumulation of NFTs and cognitive status; the spread and level of tau accumulation reflects the severity of dementia with time [61, 138-140]. The seeding activity is suggested to begin in the transentorhinal/entorhinal regions and anticipates hp-tau pathology in AD, whereas the locus ceruleus (LC) showed seeding only in later NFT stages [141]. However, immunohistochemistry has detected pre-tangle material in multiple subcortical regions, especially in locus ceruleus (LC) neurons [142-144]. Involvement of the subcortical nuclei, not considered in the original Braak scheme, however, occurs in early stages of the disease and has important clinical consequences. The cholinergic nucleus basalis of Meynert and axons of the adrenergic LC projecting neurons are affected already in Braak stages 0/I, associated with severe neuronal loss, while moderate to severe deposition of tau in the LC was only seen in Braak stages above IV [142]. The intralaminar nuclei of the thalamus, the pontine parabrachial region, the medullary reticular formation, the dorsal raphe nucleus, the oculomotor system, and the autonomous nuclei are also affected early and increase with disease progression [142, 145-147]. Nigral pathology including hp-tau (NFTs) accumulation and α-synuclein aggregates is common in elderly patients with and without AD, and may be related with extrapyramidal symptoms [148-150].
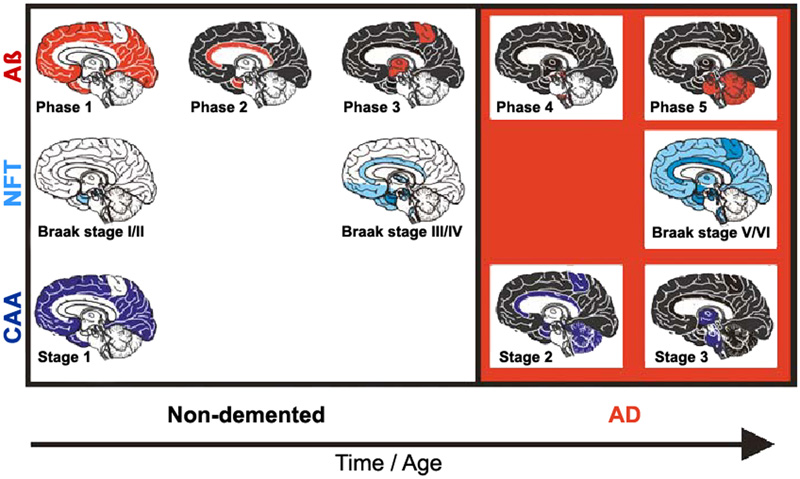
Figure 6. Spreading pattern of neuritic AD pathology. Modified from [18]. 2.8. Aβ and tau pathology - chicken or egg? The causes of sporadic AD are far from being understood, while the hallmarks that distinguish AD from other neurodegenerative diseases – namely Aβ plaques and NFTs - have been known for many years. The physiological and pathological roles of tau and Aβ, and their implications for AD pathology and therapeutics have been reviewed recently [151]. Many studies have linked Aβ and tau and raised the possibility that protein-protein interactions are the key for both spreading and toxicity of these two abnormal proteins [152]. Several models of interaction have been suggested: (1) The seeding of toxic tau is enhanced by the presence of Aβ; (2) the toxicity of Aβ depends of the presence of tau; (3) Aβ and tau enhance each other's toxicity. Modern network-based models revealed ways in which Aβ and tau protein might interact with each other to enhance the propagation of AD, thus shedding light on the importance of protein clearance and protein interaction mechanisms in the development of AD pathology [153, 154] [155]. Soluble oligomeric Aβ is hypothesized to be a possible cause of the hyperphosphorylation of tau and the development of NFTs. The presence of APs accelerates both the formation of hp-tau aggregates [156] and its interneuronal transfer [157]. The AβO hypothesis was introduced in 1998, suggesting that the brain damage leading to AD was initiated by soluble ligand-like AβOs [158]. The extension of tau pathology is different from the spread of Aβ deposition that is related to diffusion of soluble Aβ in the extracellular space [159, 160]. Quantification of ADNC in formalin-fixed post-mortem human brain tissue detected high amounts of Aβ in the frontal cortex and striatum, and of hp-tau in the frontal cortex and hippocampus of cases with high ADNC pathology load [161]. The most recent version of the amyloid cascade hypothesis assumes AD arises from synaptic toxicity mediated by soluble AβOs, leading to synaptic dysfunction and loss. Age-related aggregation of Aβ and its apparent downstream effects on microglia, astrocytes, and neurons, including the post-translational modification of the tau protein, seems necessary for AD symptom expression [162]. While an optimal concentration of Aβ is thought to likely maintain synapses, alterations in the proteolytic processing of APP may cause dyshomeostasis of Aβ, increasing the levels of Aβ-42, and initiating AD by setting off a chain of events that leads to the accumulation of tau and downstream neuronal cell death [163]. Soluble AβOs are now suggested to cause neuronal damage [76]. They are believed to insert into membranes, while others support ligand-like accumulation at particular synapses, providing a substantial molecular basis for the cause of AD [164]. Recent data support the hypothesis that Aβ enhances tau pathology through increased spreading of tau induced by PHF in vivo [165-168], and that AβOs promote tau seeding potentiating intracellular tau aggregation [169, 170]. Intraneuronal Aβ accumulation is suggested to precede tau pathology in the entorhinal cortex [171] and to interact with hippocampal and cortical tau pathology, while in the absence of Aβ tau deposition may be insufficient for the neurodegeneration process that leads to AD [172]. Many data supporting a toxic role for AβOs have backed the AβO hypothesis for AD pathogenesis, but further advances in AβO structure-function studies are needed [158]. Recent studies point to a role for exosomes in the spreading of toxic AβOs and the associated disease progression in the AD brain [173]. However, the traditional consensus of the amyloid paradigm as a singular cause of AD has been under revision, with the accumulation of new pathobiological evidence [174]. New theories suggest that various mechanisms, including prion-like spread of Aβ and tau, vasoconstrictions, growth hormone secretagogue receptor 1α (GHSR1α), and neuroinflammation, come together at a crossroad that ultimately leads to AD [11], while others suggested that extracellular Aβ and tau act in parallel and upstream of APP [175, 176]. However, recent findings have shown that the soluble form of APP binds directly to GABABR1a and modulates synaptic transmission [177], while that of Aβ aggregates do not need APP overexpression [178] but are performed by extracellular exosomes [173]. AβOs are deposited inside synaptic terminals [179], enriched in small vesicles at the presynaptic side [47], and enhance synaptic dysfunction in AD [180]. According to others, Aβ and hp-tau may develop concomitantly within synaptic terminals [181, 182] and cause abnormalities at synapses [183]. On the other hand, preclinical evidence indicates that tau pathology can progress independently of Aβ accumulation and arises downstream of genetic risk factors for AD by an aberrant metabolic pathway [184]. The argument that insoluble Aβ and tau deposits begin forming concomitantly in the cerebral cortex of AD brains would be consistent with the argument in favor of the pathogenic importance of tau deposition. Recent quantitative studies did not find regional association between Aβ-42 and insoluble tau, but a higher regional association between total Aβ-42 and soluble tau phosphorylation. This provides evidence supporting the local interplay between Aβ and soluble hp-tau in AD brains [185], and accumulating evidence suggests that both pathologies have synergistic effects. The complex Aβ-tau interaction is important for elucidating disease pathogenesis and the design of next-generation AD therapeutical trials [153]. Targeting the common epitope could be a more effective treatment strategy than targeting only Aβ or tau alone [186]. Mounting data suggest that the prion-like spreading of diffusible oligomers and other protein aggregates from cell to cell within the brain, probably through specific neuronal networks, may contribute to AD progression [128, 187]. APP overexpression is not a prerequisite for the prion-like induction of cerebral Aβ deposition that may contribute to disease progression in AD [178], and the multiple failures of previous anti-Aβ drugs may suggest that in the AD brain, the accumulation of Aβ could be secondary to an unknown 'initial disrupting event' [188]. Processing and clearance of Aβ and tau could be related to a bidirectional relationship between ADNC and autophagy [189]. Seeded templating and neurotoxicity are two of the most critical properties attributed to oligomers that have been documented for misfolded proteins in neurodegeneration [190]. It has been speculated that cellular prion protein (PrPC) is a critical player in the interplay between Aβ and tau propagation in a large group of AD cases. Pre-existing hp-tau pathology interacting with PrPC appears to be a prerequisite for Aβ function as a hp-tau pthology acceleration via PrPC [165]. Toxic tau oligomers (tauOs) and toxic oligomeric Aβ assemblies (AOs) have prionoid characteristics and are responsible for cell-to-cell spreading in the brain. Both extra- and intracellular AβOs and tauOs (not NFTs and APs) may represent novel targets of AD research and therapeutic trials [191]. Preventing soluble AβO formation and targeting their N-terminal residues with antibodies could be an attractive combined therapeutic approach [178]. Recent studies found striking patient-to-patient heterogeneity in the hyperphosphorylated species of soluble oligomeric seed-competent tau. Its seeding capacity correlates with the aggressiveness of the clinical disease, and some post-translational modification sites appeared to be associated with both seeding activity and worse clinical outcomes, suggesting that different individuals with “typical” AD have distinct biochemical features of tau that correlated with differences in the aggressiveness of clinical course [192], supporting an important causal role of tau as a driver of clinical dysfunction in AD [193]. The synergism between Aβ deposition, NFT neurodegeneration, and CAA may be a better predictor of cognitive decline or disease progression than either pathology alone [194] (Fig. 7).
Figure 7. Staging of Aβ, NFT, and CAA in non-demented (pre-AD) and demented AD patients. From [195]. 2.9. Synaptic and neuronal loss Essential neuropathological features of AD are loss of synapses and selected neuronal cells (20-40% in neocortex and 25-65% in hippocampus) as the main pathological substrate of cortical atrophy. Its regional and laminar pattern parallels the distribution of NFTs and has been suggested to be a better correlate of cognitive deficits than the Aβ burden [139, 196]. Little is known about the molecular basis of selective neuronal vulnerability in AD and the molecular pathways that lead to neurodegeneration, a key characteristic of the disease. It is the result of multiple molecular changes of interacting genes and pathways within vulnerable neurons [25]. The relationship between cellular senescence in the context of aging and AD have been reviewed recently [197]. Age-related intraneuronal aggregation of Aβ is colocalized with mitochondria and endosomes and less so with lysosomes and autophagosomes. Understanding age-related changes in intraneuronal Aβ may lead to application of countermeasures to prolong dementia-free health span [198]. The intraneuronal accumulation of Aβ may involve synaptic dysfunction and the formation of APs in AD; intraneuronal Aβ-42 has been reported to disrupt the normal cytoarchitecture of neurites. Recent studies indicate that in AD, vulnerable-neuron-specific dysregulation of polypyrimidine tract binding protein (PTB) (NCBI gene ID 5725), a regulator of alternative splicing [199], is the protein most highly correlated to tau in the principal neurons of the entorhinal cortex layer II (EC II). PTB could precipitate a 3R/4R tau imbalance in these neurons and explain the premature accumulation of NFTs, thus explaining the vulnerability of EC II neurons [25]. The neurotoxic effect of astrocyte-derived exosomes (ADE) is evident with the overlap of AP density and C3/4 fragments (complement factors) observed in early AD [200]. Although tangle-bearing neurons can be long lasting in regions where NFTs occur at a presymptomatic stage, neuronal loss occurs early in the course of the symptomatic disease [201]. Two mechanisms of neuronal death in AD have been discussed: one affecting tangle-bearing neurons that will lead to ghost extracellular tangles, another affecting tangle-free neurons, at least in part by apoptosis [202-204]. Inflammation-induced hyperphosphorylation of tau destabilizes the microtubule-actin network and impairs axonal transport and disturbs energy metabolism in the axon, inducing further tau phosphorylation. Accumulating data point to the fact that this facilitates the formation of PHFs, further impairs axonal transport leading to complete blockage and axonal leakage, and induces loss of synaptic contacts promoting activation of microglia and reactive astrogliosis [56]. Microglia have been shown to instigate tau pathology in diverse ways, inducing tau aggregation by proinflammatory cytokine release [205, 206], and spreading hp-tau oligomers or NFTs through exosome secretion [130]. Synaptic loss that is possibly driven by Aβ and tau pathology has been suggested to precede neuronal loss [207]. Synapses are present in APs and their total number decreases with time [61, 208]. Their loss has been demonstrated ultrastructurally and immunohistochemically [209]. In late stages of AD synapse loss ranges from 10 to 60%, most severely in the frontal and mesiotemporal regions. Synapse loss by activated astrocytes producing different secretomes reduce protein synthesis for synapse formation, resulting in synaptic loss found in AD [210]. There is a close relationship between Aβ accumulation and synaptic loss that may provide direction for the development of potential disease-modifying treatments of AD [211]. EOAD is associated with a higher burden of ADNC and a higher rate of neocortical atrophy and synapse loss than the much more common and apparently sporadic LOAD [212]. However, synaptic loss is not a unique hallmark of AD and occurs in many other brain diseases [213]. 2.10. Neuroinflammation Activated microglia operating as phagocytes are frequently observed around Aβ plaques driving an inflammatory response, which can be activated by multiple factors in the local environment [214], in particular by the presence of Aβ in the cortex, indicating a “toxic” response which corrupts neurons as collateral damage (“bystander effect”) [215]. Tau-positive NPs being early in AD, however, major tau deposition follows the accumulation of Aβ and clustering of activated microglia. An increase in membrane attack complex formation leads to increased tau pathology and neoronal loss [216]. On the other hand, microglia may contribute to elimination of tau deposits by phagocytosis [217, 218]. Different states of microglia activation, corresponding to regional activation of Aβ and tau, are present simultaneously in the same brain. The clustering of activated microglia is greatest in the primary motor cortex, a region relatively spared compared to the severely affected inferior temporal cortex in AD. This suggests that microglial activation is not prominent in the early phase of AD pathophysiology [66]. Recent studies in hp-tau mice demonstrated that microglia are not the agitators of tau aggregation, but different results about the involvement of microglia in tau aggregation and clearance were presented [219, 220]. Thus, the functional role of microglial activation with hp-tau oligomers still remains elusive. Gene-profiling technologies applied to isolated microglia have challenged the hypothesis that there is one acute-type (microglial drivers) of inflammation in the human brain causing accelerated proinflammatory damage in AD. These studies have shown that many of the microglia genes expressed in increased levels reflect a response to restore homeostasis and limit inflammatory damage [221]. On the other hand, there is an early microglia reaction to AD pathology, but a loss of healthy microglia is the prominent feature in severely affected regions of the AD brain [222]. In addition, there is a non-disease-specific response of microglia to neuronal damage, with upregulation of phagocytotic activity to remove damaged neurons and synapses by CD68 immunoreactivity of lysosomes [73]. Their numbers increase on promotion to neuronal damage associated with NFTs [62], which is due to enhanced production of inflammatory cytokines, such as IL-21 and increase in T follicular helper cells. The strong immune response is insufficient at clearing up Aβ and instead exacerbates inflammation [223]. Reactive astrocytes that may react to cytokines and other agents produced by pro-inflammatory microglia, are observed around APs, though less frequently compared to microglia. Reactive astroglia burden occurs later in AD and correlates mainly with tau pathology [31]. 2.11. Pathology of preclinical AD Amyloid and neuritic plaques and NFTs occurring in non-demented elderly individuals, represent asymptomatic or preclinical AD (pre-AD), while clinical AD affects subjects with late stages of ADNC. Both AD and pre-AD cases often exhibit CAA, which is also observed in non-AD cases, i.e., those without ADNC. Patients with MCI do not always have ADNC even though they have a risk of developing dementia in 10-12% and sometimes do not have any discernable pathology [224, 225]. The presence of NFTs and CAA in cases without APs, classified as non-AD, suggests that they may precede AP pathology or may present a pre-amyloid plaque stage not yet included in the current criteria for the neuropathological diagnosis of AD [226]. Increased soluble/dispersible Aβ in pre-AD compared to fully developed suggests that, in addition to more severe and widespread ADNC, soluble Aβ aggregates play a role in the conversion of pre-AD to clinical AD [31]. Cognitively impaired individuals presenting with an early onset AD phenotype showed higher rates of tau PET accumulation, while among cognitively unimpaired individuals higher rates of tau accumulation were associated with faster rates of memory decline [227]. 2.12. Neuropathological diagnosis of Alzheimer’s disease
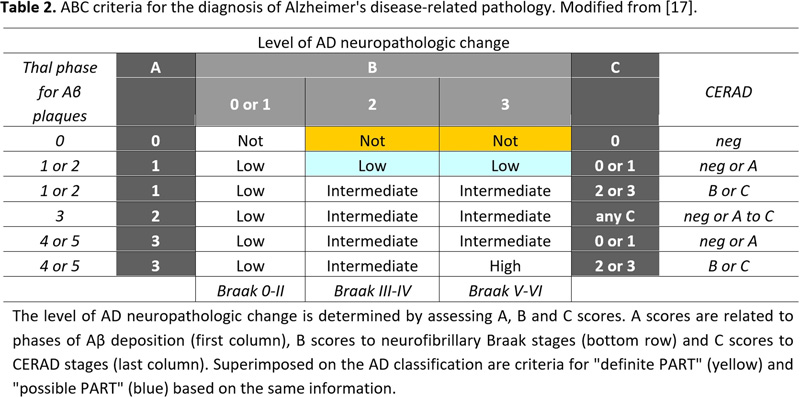
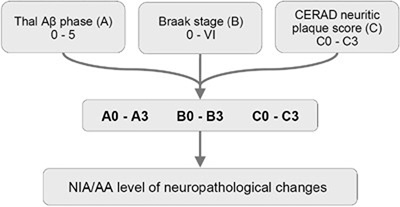
Histopathological examination of the brain has to establish that ADNC are present in sufficient densities and extensions to distinguish AD from other age-related disorders [61]. Because the disease affects the whole brain, it is not sufficient to make the diagnosis of AD just on one or two brain blocks; instead, multiple brain areas have to be examined and a staging protocol has to be established. The current algorithms for the pathological diagnosis of AD are based on semiquantiative assessment of APs and NFTs providing reasonable interrater agreement when using standardized criteria [228]. Current guidelines include (a) cut-off quantitative values for APs and tangles [17, 229]; (b) the semiquantitative assessment and age-adjustment of NPs in the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) protocol [230]; (c) topographic staging of neuritic/tau pathology [18], re-evaluated by immunohistochemistry [231]; and (d) the progress and distribution of Aβ phases [79]. In order to develop a system that combines all the above pathological features, the NIA/AA established a composite score comprising the extent of involvement/spread of cerebral Aβ based on the progression model by the Thal phases: (A), that of NFTs based on the progression model of Braak, (B), and the CERAD score, which describes the density of neuritic amyloid plaques based on certain key locations in the neocortex, (C) (Table 1). From this combination, it gave a likelihood for the degree of AD neuropathological changes in an individual case. Sufficient agreement in AD diagnosis could be reached only when the lesions are considerable (Braak NFT stage V and VI) with 91% agreement, while for mild lesions it was poorer (for Braak stage I and II, agreement was only around 50%) [228, 232, 233].
Figure 8. Pathway of the combination of different pathological features that allows a classification of ADNC according to the NIA-AA guidelines. A comparative study of clinical and neuropathological diagnoses of AD in three epidemiological samples reported a sensitivity for probable AD of 93% [238]. Meta-analysis of 20 (out of 1,189) studies to distinguish autopsy-verified AD from other dementias or healthy controls showed a sensitivity of 85.4% (95% CI 80.8-90%) and a specificity of 77.7% (95% CI 70.2-85.1%). Values were higher for neuroimaging procedures and slightly lower for CSF biomarkers, while the combination of both resulted in better results [239]. 3. Pathobiological subtypes of AD
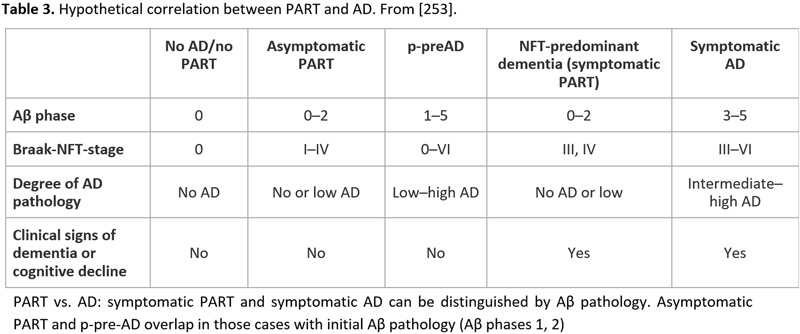
Recent studies showed that the neuropathology of AD is heterogenous [240-242]. The current guidelines for the neuropathological diagnosis of AD only consider the classical “plaque and tangle” phenotype but not other subtypes such as the “plaque only but without tangle formation/predominant” type with abundant amyloid, or the “little or no tau pathology” type limited to the hippocampus and abnormal hp-tau in neocortical pyramidal cells. This type, observed in 3.4-8.0% of demented subjects over age 85 years [243], frequently represents a specific type of dementia with Lewy bodies (DLB)/DLB-AD [244]. The recently described “primary age-related taupathy” (PART) [245], previously referred to as “NFT-predominant dementia” [246], involves people over 85 years old and is associated with mild to moderate cognitive impairment [247, 248] It reveals tau pathology restricted to the MTL (Braak stages 0-IV), relative absence of amyloid (Thal Aβ phases 0-2), total absence of NPs, and rare CAA [249]. The composition of NFTs in PART both for 3R and 4R tau isoforms is identical with those in classical AD [246], while pattern of hippocampal tau pathology differs significantly between PART and AD [250, 251]. Tau aggregates influence cognition and hippocampal atrophy in the absence of Aβ [249]. Positive correlations were reported in PART between the Braak NFT stage and phosphorylated 43-kDa TAR DNA-binding protein (pTDP-43) stage and density [252]. PART is considered either a prodromal form or a subtype of AD [253, 254] (see Table 3). MAPT H1H1 genotype frequency is high in both PART and limbic-predominant AD (LP-AD), and similar to typical AD, while APOEε4 is rather rare in PART [255]. Other genetic differences between PART and AD have been described [256]. It seems that lower concentrations of AβOs cause less severe tau deposition due to the fact that they can potentiate tau aggregation by promoting tau seed uptake [170]. Cognitive decline in PART is usually milder than in AD and correlates with tau burden. Biomarkers and neuroimaging studies will be important to define PART ante-mortem and to follow its natural history [257]. While the incidence of classical AD increases from the 7th to the 9th decade and later shows a mild decrease, the frequency of PART increases after the age of 85 years [3].
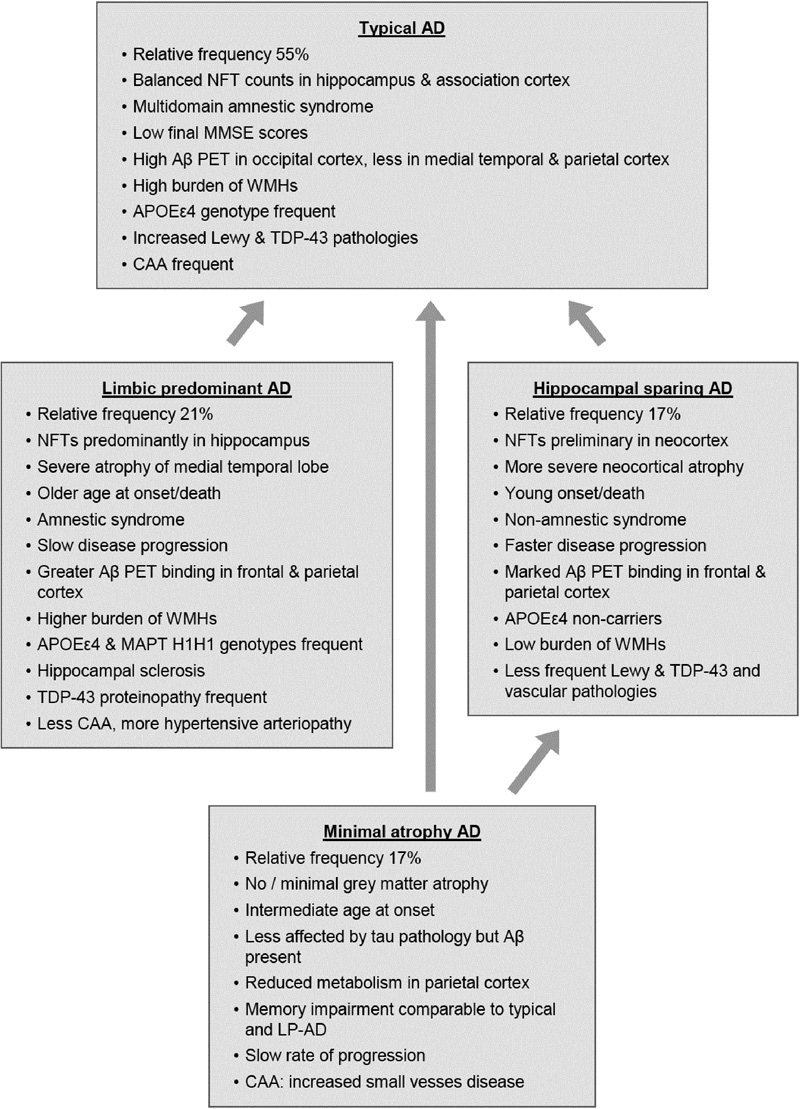
Recent clinicopathological studies have enabled the identification of several pathophysiologically defined subtypes of AD. One distinguished three AD subtypes based on NFT density: typical AD with balanced NFT counts in the neocortex and hippocampus (75%), hippocampal sparing (HcSp), with NFT counts predominantly in association cortices (11%), and limbic-predominant (LP) AD mainly involving the hippocampus (14%) [262]. These subtypes had different clinical phenotypes, with different ages at onset and rates of progression (Fig. 9). Patients with hippocampal sparing AD (HcSp-AD) were youngest at onset, had a higher proportion of men, and progressed more quickly than typical AD. LP-AD patients were older, more often female, and showed slower progression. Age at death of the LP form was highest, while patients with HcSp-AD were youngest, indicating this type as the most aggressive. This could be related to the contribution of TDP-43 pathology, hippocampal sclerosis, and the microtubule-associated protein tau (MAPT) H1H1 genotype to LP-AD, factors related to temporal lobe atrophy, older age, and slower disease progression. APOEε4 carriers more frequently had LP-AD and typical AD, whereas non-carriers more frequently presented as HcSp-AD. Vascular co-pathology (ranging from 16 to 36%) was highest in the LP and lowest in the HcSp cases. Typical AD had higher AP burden in occipital regions compared with LP-AD [262], while in contrast to specific tau accumulation and brain atrophy patterns among AD variants, Aβ accumulation appeared rather diffuse and similarly across groups, except the MA group [263, 264]. Tau pathology was closely associated with sites of neurodegeneration and brain atrophy corresponded well with NFT topography and neuronal loss. [265-267]. Clinical symptoms correlate with neuronal hypometabolism [262, 268, 269]. Similar results were reported in a study of 933 autopsy cases of AD, all with neuritic Braak stage > IV [270]. Typical AD was more frequent than in the Mayo series (82.5 vs 75%), while the other two subtypes were slightly less frequent. Minimal-atrophy AD (MA-AD) was not included in this study. The LP-AD cases shared some morphological features with PART [245], although later studies demonstrated significant pathological differences between PART and LP-AD [240].
Figure 9. Main factors and characteristics of the four major subtypes of AD. AD: Alzheimer’s disease; NFT: neurofibrillary tangle; WMH: white matter hyperintensity; CAA: cerebral amyloid angiopathy; EOAD: early-onset Alzheimer’s disease; LOAD: late-onset Alzheimer’s disease; LP-AD: limbic-predominant AD. Typical AD showed greater white matter hyperintensity (WMH) burden, which may be due to Wallerian degeneration induced by cortical tau pathology [271, 272], small vessel disease, or both [273-275]. Tau pathology and neurodegeneration can disrupt key brain networks, which may induce memory impairment comparable to LP-AD and typical AD in the absence of overt brain atrophy of the MTL in MA-AD that shows [276-278]. Distinct patterns of NFT deposition in young-onset versus older-onset AD give evidence for variability in regional deposition patterns and demonstrate that different disease phenotypes have different patterns of tau pathology [279]. Other atypical non-amnestic syndromes, referred to as focal AD [26, 280, 281], include logopenic primary progressive aphasia (LPPA), showing higher NFT density in superior temporal gyrus but Thal amyloid plaques similar to amnestic AD [282-284]. The proportion of APOEε4 carriers was elevated in amnestic but not in non-amnestic forms of AD, suggesting that APOE is a selective risk factor that increases the vulnerability of memory-related medial temporal areas rather than language-related neocortices [285]. Further atypical forms are posterior cortical atrophy (PCA) [286], non-amnestic AD with TDP-43 pathology [287], syndromes resembling behavioral variant fronto-temporal lobe degeneration with tau pathology (bvFTD-tau) [266, 288], and the corticobasal syndrome (CBS) subtype of AD that shows a higher NFT density in the perirolandic cortices and greater neuronal loss in substantia nigra which may contribute to parkinsonism that uncommon in classic AD [289]. Behavioral/dysexecutive AD revealed temporo-parietal-predominant atrophy. In the Mayo series, PCA, LPPA and bvFTD variants were more common in HcSp-AD than in LP and typical AD [262, 290]. Accumulation of NFTs and activated hypertrophic microglia associated with low neuron densities suggests that they may collectively contribute to focal neurodegeneration characteristic of primary progressive aphasia AD [291]. The pathogenic factors underlying AD subtypes are unclear and cannot be explained by Aβ pathology alone, because the distribution of Aβ PET retention is quite similar in all subtypes [263]. However, solid-state nuclear magnetic resonance measurements showed qualitative differences between Aβ-40 and Aβ-42 aggregates in the brain tissue of patients with two atypical AD clinical subtypes - posterior cortical atrophy variant and a typical prolonged-duration form - indicating that there are structural variations in Aβ fibrils from AD clinical subtypes [60]. MA-AD, although Aβ-positive, shows less tau pathology. According to recent studies, AD “subtypes” may be linked to different tau protein modifications, suggesting that AD patients may have multiple molecular drivers of an otherwise common phenotype [192]. This suggests that multiple subtypes are parts of the same AD continuum [266], which may have consequences for personalized therapeutic approaches. 4. The impact of co-pathologies AD pathology rarely occurs in isolation, while complex pathologies frequently lead to cognitive decline. The number of co-morbidities increases in the aging brain, causing mixed pathologies [3, 248, 274, 292-298]. The challenges of pathological mimics and concomitant pathologies in the neuropathologic diagnosis of AD have been critically reviewed recently [174]. The most frequent co-pathologies are cerebrovascular disease (CVD) and Lewy and TDP-43 proteinopathies [31, 258, 299, 300]. In a consecutive autopsy series of 2,060 elderly demented patients and those with the clinical diagnosis of AD, ADNC were present in 82.9% of all demented and in 92.8% of clinically diagnosed AD cases, but only 33.6% and 47.6%, respectively, showed pure ADNC (ABC 3/3/3). The others were either atypical AD forms or subtypes (including PART) (7 and 6%, respectively) or exhibited additional CVD (24.3%), Lewy (12.5%) or other mixed pathologies. Vascular dementia in this cohort accounted for only 12.2% and 3.3%, respectively; other non-AD pathologies were present in 7.2% and 3.7% [301]. Another study of demented elderly persons reported pure ADNC in only 31% and multiple pathologies in 63% [302]. A review of 12 studies with 3,574 patients, irrespective of the clinical symptoms, reported ADNC between 19% and 67%, Lewy pathologies in 6% to 39%, vascular pathologies in 28% and 70%, TDP-43 proteinopathy in 19% to 78% [290], hippocampal sclerosis between 3% and 13%, and mixed pathologies between 8% and 70% [295]. Among 447 patients with probable AD, only 3.13% showed pure ADNC, 27.3% AD+CVD + other, 3% AD + CVD, 7.6% AD + other degenerative lesions, and 47% AD+CVD + other neurodegenerative lesions [293]. This list of combinations is not complete and there are other combinations with rare entities that need specific attention [174]. Among 673 autopsy cases, including 320 demented, the majority showed mixed pathologies [274]. LATE-NC was present in 57% of AD cases and was associated with more rapid disease progression [259]. Increased TDP-43 pathology in typical AD and LP-AD compared to HcSp-AD [262] was due to a strong association between hippocampal sclerosis and TDP-43, but clinical presentation seemed to be driven by morphological subtypes and not by TDP-43 pathology [290]. Among 61 autopsy-proven AD cases, LATE-NC was present in 67.2% (AD, LATE-NC), however, it was not associated with an increase of the burden of early or late tau nor Aβ pathology. LATE-NC showed a lower final mini mental state examination (MMSE) score independent of tau pathology [303]. Among 172 autopsy-confirmed AD cases, 19% were classified as non-amnestic, 69% of which had typical ADNC, 31% were HcSp-AD, 36% TDP-43-positive, while there were no LP-AD cases [287]. In a recent study of 46 autopsy-confirmed AD cases, 63% exhibited LATE-NC (AD+) and a higher burden of hp-tau. This indicates a possible progression of the disease, whereas AD plus LATE-NC was not associated with differences in cognitive scores [304]. LATE-NC may also occur in isolation and has been viewed as a common brain disease in aging [258]. Among 574 individuals with complete measurements of MMSE and the Clinical Dementia Rating scale sum of boxes (CDR-SB) from 39 AD centers across the USA, 63% of those given the 'gold standard' diagnosis of AD, possessed either TDP-43 proteinopathy or CAA of sufficient severity to independently explain the majority of their cognitive impairment. Aβ and/or tau burden, particularly in Braak stages IV to VI, and small cerebral vessel disease may synergistically affect cognitive decline [305], and a significant interaction was found between Braak NFT stages, CAA status and cognitive decline, suggesting that there is a significant interaction between tau pathology and CAA on cognition within the AD clinical spectrum [306]. Hence, interventions targeting CAA may contribute to delay the onset of cognitive impairment, particularly in individuals with intermediate ADNC [307]. This suggests that many individuals diagnosed with AD may actually suffer from a mixed dementia, and therapeutic targeting AD-related processes only may have limited efficiency in these co-morbid populations [298]. Based on data from the NACC, 1,854 participants with a clinical diagnosis of AD and ADNC at autopsy (confirmed AD) were studied; 204 with the clinical AD diagnosis had no ADNC (AD-mimics), while 253 participants with negative clinical AD diagnosis had ADNC (unidentified AD). Compared to confirmed AD cases, AD-mimics (FTLD-tau, hippocampal sclerosis, cerebrovascular pathology, etc.) had less severe cognitive impairment [308]. Special practical considerations for the diagnosis of essential co-pathologies and their relations with AD were given recently [174]. Argyrophilic grain disease (AGD), a limbic-predominant 4R-tauopathy, with grain-like deposits in neuritic dendrites, oligodendroglial inclusions (“coiled bodies”), ramified astrocytes, and ballooned neurons in the amygdala, hippocampus and MTLs [309], represents an age-related disorder and has been reported in up to 25% AD cases [310], and rarely occurs before the age of 75 [311]. Aging-related tau astrogliopathy (ARTAG) is defined by the presence of two types of tau-bearing astrocytes: thorn-shaped and granular/fuzzy astrocytes in the brains of old-aged individuals in different locations and anatomical regions (subependymal, subpial, perivascular, white and gray matter [312, 313]. Among additional pathological changes in AD is granulovacuolar degeneration (GVD), characterized as 3-5 μm vesicles bound by a unit membrane, most frequently occurring in the pyramidal neurons of the hippocampus, usually in association with NFTs. Their origin and significance are unclear. Despite the strong association between tau aggregation and granulovacuolar degeneration body (GVB) formation [314], intracellular aggregates of proteins other than tau can also induce GVB formation, which needs further elucidation [315]. The granule of the GVD is immunolabeled by antibodies against tubulin, ubiquitin, neurofilament, and tau [31]. They correlate with NFT density, suggesting that they may be a cellular response to neuronal damage or late-stage autophagic vacuoles [316]. Necrosome complex detected in GVD is associated with neuron loss in AD [317]. Recent clinicopathological studies have shown that the complex cascades of the underlying pathologies in most elderly patients may lead to cognitive decline, and that the number of possible combinations due to co-morbidities increases with aging [248]. These concomitant pathologies may be harmful to individuals with low cognitive reserve such as patients with MA-AD. They can cause a number of challenges including the evaluation of the significance of each pathological entity in the manifestation of the clinical symptoms, and the threshold of each individual pathology to cause dementia [174]. Total burden of comorbid pathological abnormalities, rather than any single lesion, is the most important cause of cognitive impairment, often despite clinical diagnosis of “only” AD [318]. 5. Conclusions AD is a heterogeneous, multifactorial disorder, manifesting clinically and morphologically as several subtypes that have a distinct signature of network disruptions associated with their atrophy pattern and reflecting the differential spread of NFT pathology and neuronal loss due to different vulnerability patterns of affected brain regions, which relates to specific molecular-functional properties of the affected neuronal systems [22, 23]. The severity of lesions corresponds to the “N” category in the new A/T/N classification for biomarkers [319]. The heterogeneity of the Alzheimer’s syndrome is related to multiple pathogenic factors which induce misfolding tau, Aβ, TDP-43, and other proteins, the synergetic or additive action of which results in various disease phenotypes [320]. Several factors such as brain resilience may help compensate for these pathologies up to a certain level, although their relevance is still poorly understood. These problems and the increasing incidence of AD illustrate its consequences on public health and the resulting challenges for future medicine. Increased sensitivity and specificity of new ATN biomarker systems and more extensive clinicopathological studies in well-defined populations are needed, with post-mortem studies using the updated NIA/AA criteria. The recent advent of tau PET and novel imaging and fluid-based (CSF) biomarkers allows us to study the temporal progression of tau pathology in vivo [321, 322]. Improving methods for disease detection and monitoring its progression may hopefully lead to the development and refinement of tau-based therapeutics. In the interest of optimizing the clinical diagnosis of AD and related disorders, neuropathological studies should use a wide range of molecular pathological methods and should evaluate multiple CNS regions. An optimal and less cost-intensive strategy would be to screen specifically neurodegeneration-related proteins and to examine their cross reactions. The recent correlative work on concomitant pathologies has provide insight into the interactions of the various pathologies and their roles in causing dementing symptoms. Interdisciplinary studies may improve our knowledge about the pathogenesis of the heterogeneous manifestation of AD and promote methods for its early diagnosis as the basis for further preventive and successful disease-modifying therapeutic measures. Funding This research was partially funded by the Society for the Promotion of Research in Experimental Neurology, Vienna, Austria. Acknowledgments The author thanks Mr. E. Mitter-Ferstl, PhD, for secretarial and editorial work. Conflicts of Interest The author declares no conflict of interest. References 1. Alzheimer's-Association (2018) 2018 Alzheimer's disease facts and figures. Alzheimers Dement 14:367-429 2. Farfel JM, Yu L, Boyle PA, Leurgans S, Shah RC, Schneider JA, Bennett DA (2019) Alzheimer's disease frequency peaks in the tenth decade and is lower afterwards. Acta Neuropathol Commun 7:104 3. Jellinger KA, Attems J (2010) Prevalence of dementia disorders in the oldest-old: an autopsy study. Acta Neuropathol 119:421-433 4. Fiest KM, Roberts JI, Maxwell CJ, Hogan DB, Smith EE, Frolkis A, Cohen A, Kirk A, Pearson D, Pringsheim T, Venegas-Torres A, Jetté N (2016) The prevalence and incidence of dementia due to Alzheimer's disease: a systematic review and meta-analysis. Canad J Neurol Sci 43 (Suppl S1):S51-S82 5. Alzheimer's-Association (2020) 2020 Alzheimer's disease facts and figures. Alzheimers Dement online Mar 10: doi: 10.1002/alz.12068 6. Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP (2013) The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement 9:63-75 e62 7. Long JM, Holtzman DM (2019) Alzheimer disease: an update on pathobiology and treatment strategies. Cell 179:312-339 8. Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC (2004) Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 62:925-931 9. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M, Amit I (2017) A unique microglia type associated with restricting development of Alzheimer's disease. Cell 169:1276-1290 e1217 10. Monzio Compagnoni G, Di Fonzo A, Corti S, Comi GP, Bresolin N, Masliah E (2020) The role of mitochondria in neurodegenerative diseases: the lesson from Alzheimer's disease and Parkinson's disease. Mol Neurobiol 57:2959-2980 11. Gallardo G, Holtzman DM (2019) Amyloid-beta and tau at the crossroads of Alzheimer's disease. Adv Exp Med Biol 1184:187-203 12. Kisler K, Nelson AR, Montagne A, Zlokovic BV (2017) Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci 18:419-434 13. Nation DA, Sweeney MD, Montagne A, Sagare AP, D'Orazio LM, Pachicano M, Sepehrband F, Nelson AR, Buennagel DP, Harrington MG, Benzinger TLS, Fagan AM, Ringman JM, Schneider LS, Morris JC, Chui HC, Law M, Toga AW, Zlokovic BV (2019) Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med 25:270-276 14. Carter SF, Herholz K, Rosa-Neto P, Pellerin L, Nordberg A, Zimmer ER (2019) Astrocyte biomarkers in Alzheimer's disease. Trends Mol Med 25:77-95 15. Jack CR, Jr., Therneau TM, Weigand SD, Wiste HJ, Knopman DS, Vemuri P, Lowe VJ, Mielke MM, Roberts RO, Machulda MM, Graff-Radford J, Jones DT, Schwarz CG, Gunter JL, Senjem ML, Rocca WA, Petersen RC (2019) Prevalence of biologically vs clinically defined Alzheimer spectrum entities using the National Institute on Aging-Alzheimer's Association Research Framework. JAMA Neurol 76:1174-1183 16. Ferreira D, Nordberg A, Westman E (2020) Biological subtypes of Alzheimer disease: A systematic review and meta-analysis. Neurology 94:436-448 17. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters HV, Hyman BT (2012) National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 123:1-11 18. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239-259 19. Fornari S, Schäfer A, Jucker M, Goriely A, Kuhl E (2019) Prion-like spreading of Alzheimer's disease within the brain's connectome. J R Soc Interface 16:20190356 20. Mattsson N, Schott JM, Hardy J, Turner MR, Zetterberg H (2016) Selective vulnerability in neurodegeneration: insights from clinical variants of Alzheimer's disease. J Neurol Neurosurg Psychiatry 87:1000-1004 21. Mrdjen D, Fox EJ, Bukhari SA, Montine KS, Bendall SC, Montine TJ (2019) The basis of cellular and regional vulnerability in Alzheimer's disease. Acta Neuropathol 138:729-749 22. Grothe MJ, Sepulcre J, Gonzalez-Escamilla G, Jelistratova I, Schöll M, Hansson O, Teipel SJ (2018) Molecular properties underlying regional vulnerability to Alzheimer's disease pathology. Brain 141:2755-2771 23. Wang ZT, Zhang C, Wang YJ, Dong Q, Tan L, Yu JT (2020) Selective neuronal vulnerability in Alzheimer's disease. Ageing Res Rev 62:101114 24. Acosta D, Powell F, Zhao Y, Raj A (2018) Regional vulnerability in Alzheimer's disease: The role of cell-autonomous and transneuronal processes. Alzheimers Dement 14:797-810 25. Roussarie JP, Yao V, Rodriguez-Rodriguez P, Oughtred R, Rust J, Plautz Z, Kasturia S, Albornoz C, Wang W, Schmidt EF, Dannenfelser R, Tadych A, Brichta L, Barnea-Cramer A, Heintz N, Hof PR, Heiman M, Dolinski K, Flajolet M, Troyanskaya OG, Greengard P (2020) Selective neuronal vulnerability in Alzheimer's disease: a network-based analysis. Neuron 107:P821-835.e812 26. Warren JD, Fletcher PD, Golden HL (2012) The paradox of syndromic diversity in Alzheimer disease. Nat Rev Neurol 8:451-464 27. Jellinger KA (2020) Towards a biological definition of Alzheimer disease. Int J Neurol Neurother 7:095; DOI 010.23937/22378-23001/1410095 28. Zhou J, Greicius MD, Gennatas ED, Growdon ME, Jang JY, Rabinovici GD, Kramer JH, Weiner M, Miller BL, Seeley WW (2010) Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer's disease. Brain 133:1352-1367 29. Piguet O, Double KL, Kril JJ, Harasty J, Macdonald V, McRitchie DA, Halliday GM (2009) White matter loss in healthy ageing: a postmortem analysis. Neurobiol Aging 30:1288-1295 30. Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1:a006189 31. DeTure MA, Dickson DW (2019) The neuropathological diagnosis of Alzheimer's disease. Mol Neurodegener 14:32 32. Tome SO, Vandenberghe R, Ospitalieri S, Van Schoor E, Tousseyn T, Otto M, von Arnim CAF, Thal DR (2020) Distinct molecular patterns of TDP-43 pathology in Alzheimer's disease: relationship with clinical phenotypes. Acta Neuropathol Commun 8:61 33. O'Brien RJ, Wong PC (2011) Amyloid precursor protein processing and Alzheimer's disease. Annu Rev Neurosci 34:185-204 34. Goldgaber D, Lerman MI, McBride OW, Saffiotti U, Gajdusek DC (1987) Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer's disease. Science 235:877-880 35. Tanzi RE, Gusella JF, Watkins PC, Bruns GA, St George-Hyslop P, Van Keuren ML, Patterson D, Pagan S, Kurnit DM, Neve RL (1987) Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science 235:880-884 36. Coronel R, Bernabeu-Zornoza A, Palmer C, Muniz-Moreno M, Zambrano A, Cano E, Liste I (2018) Role of amyloid precursor protein (APP) and its derivatives in the biology and cell fate specification of neural stem cells. Mol Neurobiol 55:7107-7117 37. Zheng H, Koo EH (2011) Biology and pathophysiology of the amyloid precursor protein. Mol Neurodegener 6:27 38. Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW, Kremmer E, Rossner S, Lichtenthaler SF (2010) ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J 29:3020-3032 39. Postina R, Schroeder A, Dewachter I, Bohl J, Schmitt U, Kojro E, Prinzen C, Endres K, Hiemke C, Blessing M, Flamez P, Dequenne A, Godaux E, van Leuven F, Fahrenholz F (2004) A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J Clin Invest 113:1456-1464 40. Kojro E, Fahrenholz F (2005) The non-amyloidogenic pathway: structure and function of alpha-secretases. Subcell Biochem 38:105-127 41. Nhan HS, Chiang K, Koo EH (2015) The multifaceted nature of amyloid precursor protein and its proteolytic fragments: friends and foes. Acta Neuropathol 129:1-19 42. De Chiara G, Marcocci ME, Civitelli L, Argnani R, Piacentini R, Ripoli C, Manservigi R, Grassi C, Garaci E, Palamara AT (2010) APP processing induced by herpes simplex virus type 1 (HSV-1) yields several APP fragments in human and rat neuronal cells. PLoS One 5:e13989 43. Portelius E, Brinkmalm G, Tran A, Andreasson U, Zetterberg H, Westman-Brinkmalm A, Blennow K, Ohrfelt A (2010) Identification of novel N-terminal fragments of amyloid precursor protein in cerebrospinal fluid. Exp Neurol 223:351-358 44. Frost GR, Li YM (2017) The role of astrocytes in amyloid production and Alzheimer's disease. Open Biol 7 45. Peters F, Salihoglu H, Rodrigues E, Herzog E, Blume T, Filser S, Dorostkar M, Shimshek DR, Brose N, Neumann U, Herms J (2018) BACE1 inhibition more effectively suppresses initiation than progression of beta-amyloid pathology. Acta Neuropathol 135:695-710 46. Volloch V, Olsen B, Rits S (2020) Alzheimer's disease is driven by intraneuronally retained beta-amyloid produced in the AD-specific, beta-app-independent pathway: current perspective and experimental models for tomorrow. Ann Integr Mol Med 2:90-114 47. Yu Y, Jans DC, Winblad B, Tjernberg LO, Schedin-Weiss S (2018) Neuronal Abeta42 is enriched in small vesicles at the presynaptic side of synapses. Life Sci Alliance 1:e201800028 48. Greenfield JP, Tsai J, Gouras GK, Hai B, Thinakaran G, Checler F, Sisodia SS, Greengard P, Xu H (1999) Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc Natl Acad Sci U S A 96:742-747 49. Hartmann T, Bieger SC, Brühl B, Tienari PJ, Ida N, Allsop D, Roberts GW, Masters CL, Dotti CG, Unsicker K, Beyreuther K (1997) Distinct sites of intracellular production for Alzheimer's disease A beta40/42 amyloid peptides. Nat Med 3:1016-1020 50. Hellström-Lindahl E, Viitanen M, Marutle A (2009) Comparison of Abeta levels in the brain of familial and sporadic Alzheimer's disease. Neurochem Int 55:243-252 51. Walsh DM, Selkoe DJ (2007) A beta oligomers - a decade of discovery. J Neurochem 101:1172-1184 52. Galvao F, Jr., Grokoski KC, da Silva BB, Lamers ML, Siqueira IR (2019) The amyloid precursor protein (APP) processing as a biological link between Alzheimer's disease and cancer. Ageing Res Rev 49:83-91 53. Pinnix I, Ghiso JA, Pappolla MA, Sambamurti K (2013) Major carboxyl terminal fragments generated by gamma-secretase processing of the Alzheimer amyloid precursor are 50 and 51 amino acids long. Am J Geriatr Psychiatry 21:474-483 54. Kametani F (2008) Epsilon-secretase: reduction of amyloid precursor protein epsilon-site cleavage in Alzheimer's disease. Curr Alzheimer Res 5:165-171 55. Viswanathan J, Haapasalo A, Kurkinen KM, Natunen T, Mäkinen P, Bertram L, Soininen H, Tanzi RE, Hiltunen M (2013) Ubiquilin-1 modulates gamma-secretase-mediated epsilon-site cleavage in neuronal cells. Biochemistry 52:3899-3912 56. Krstic D, Knuesel I (2013) Deciphering the mechanism underlying late-onset Alzheimer disease. Nat Rev Neurol 9:25-34 57. Michno W, Wehrli P, Meier SR, Sehlin D, Syvänen S, Zetterberg H, Blennow K, Hanrieder J (2020) Chemical imaging of evolving amyloid plaque pathology and associated Abeta peptide aggregation in a transgenic mouse model of Alzheimer's disease. J Neurochem 152:602-616 58. Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL (2015) Alzheimer's disease. Nat Rev Dis Primers 1:15056 59. Sil S, Hu G, Liao K, Niu F, Callen S, Periyasamy P, Fox HS, Buch S (2020) HIV-1 Tat-mediated astrocytic amyloidosis involves the HIF-1alpha/lncRNA BACE1-AS axis. PLoS Biol 18:e3000660 60. Qiang W, Yau WM, Lu JX, Collinge J, Tycko R (2017) Structural variation in amyloid-beta fibrils from Alzheimer's disease clinical subtypes. Nature 541:217-221 61. Duyckaerts C, Delatour B, Potier MC (2009) Classification and basic pathology of Alzheimer disease. Acta Neuropathol 118:5-36 62. Serrano-Pozo A, Mielke ML, Gomez-Isla T, Betensky RA, Growdon JH, Frosch MP, Hyman BT (2011) Reactive glia not only associates with plaques but also parallels tangles in Alzheimer's disease. Am J Pathol 179:1373-1384 63. Thal DR, Capetillo-Zarate E, Del Tredici K, Braak H (2006) The development of amyloid beta protein deposits in the aged brain. Sci Aging Knowledge Environ 2006:re1 64. Moro ML, Phillips AS, Gaimster K, Paul C, Mudher A, Nicoll JAR, Boche D (2018) Pyroglutamate and Isoaspartate modified Amyloid-Beta in ageing and Alzheimer's disease. Acta Neuropathol Commun 6:3 65. Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, Vlachouli C, Wilkinson D, Bayer A, Games D, Seubert P, Schenk D, Holmes C (2006) Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol 65:1040-1048 66. Paasila P, Davies D, Sutherland G, Goldsbury C (2020) Clustering of activated microglia occurs before the formation of dystrophic neurites in the evolution of Abeta plaques in Alzheimer’s disease. Free Neuropathol 1:20 doi 10.17879/freeneuropathology-12020-12845 67. Reed-Geaghan EG, Croxford AL, Becher B, Landreth GE (2020) Plaque-associated myeloid cells derive from resident microglia in an Alzheimer's disease model. J Exp Med 217 68. Yeh FL, Hansen DV, Sheng M (2017) TREM2, microglia, and neurodegenerative diseases. Trends Mol Med 23:512-533 69. Han S, Kollmer M, Markx D, Claus S, Walther P, Fändrich M (2017) Amyloid plaque structure and cell surface interactions of beta-amyloid fibrils revealed by electron tomography. Sci Rep 7:43577 70. Boon BDC, Bulk M, Jonker AJ, Morrema THJ, van den Berg E, Popovic M, Walter J, Kumar S, van der Lee SJ, Holstege H, Zhu X, Van Nostrand WE, Natté R, van der Weerd L, Bouwman FH, van de Berg WDJ, Rozemuller AJM, Hoozemans JJM (2020) The coarse-grained plaque: a divergent Abeta plaque-type in early-onset Alzheimer's disease. Acta Neuropathol online Sep 14: doi: 10.1007/s00401-00020-02198-00408 71. Attems J, Jellinger KA (2013) Neuropathology. In: Dening T, Thomas A (eds) Oxford Textbook of Old Age Psychiatry (2 ed.). Oxford Univ. Press Oxford, UK, pp 87-105, DOI: 110.1093/med/9780199644957.9780199644003.9780199640006 72. Oide T, Kinoshita T, Arima K (2006) Regression stage senile plaques in the natural course of Alzheimer's disease. Neuropathol Appl Neurobiol 32:539-556 73. Boche D, Nicoll JAR (2020) Understanding cause and effect in Alzheimer's pathophysiology: implications for clinical trials. Neuropathol Appl Neurobiol Jul 8:https://doi.org/10.1111/nan.12642 74. Harris JA, Devidze N, Verret L, Ho K, Halabisky B, Thwin MT, Kim D, Hamto P, Lo I, Yu GQ, Palop JJ, Masliah E, Mucke L (2010) Transsynaptic progression of amyloid-beta-induced neuronal dysfunction within the entorhinal-hippocampal network. Neuron 68:428-441 75. Yamamoto K, Tanei ZI, Hashimoto T, Wakabayashi T, Okuno H, Naka Y, Yizhar O, Fenno LE, Fukayama M, Bito H, Cirrito JR, Holtzman DM, Deisseroth K, Iwatsubo T (2015) Chronic optogenetic activation augments Abeta pathology in a mouse model of Alzheimer disease. Cell Rep 11:859-865 76. Reiss AB, Arain HA, Stecker MM, Siegart NM, Kasselman LJ (2018) Amyloid toxicity in Alzheimer's disease. Rev Neurosci 29:613-627 77. Yu L, Petyuk VA, Tasaki S, Boyle PA, Gaiteri C, Schneider JA, De Jager PL, Bennett DA (2019) Association of cortical beta-amyloid protein in the absence of insoluble deposits with Alzheimer disease. JAMA Neurol 76:818-826 78. Katzmarski N, Ziegler-Waldkirch S, Scheffler N, Witt C, Abou-Ajram C, Nuscher B, Prinz M, Haass C, Meyer-Luehmann M (2020) Abeta oligomers trigger and accelerate Abeta seeding. Brain Pathol 30:36-45 79. Thal DR, Rub U, Orantes M, Braak H (2002) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58:1791-1800 80. Walker LC (2020) Aß plaques. Free Neuropathol 1:31, DOI 10.17879/freeneuropathology-12020-13025 81. Thal DR, Ronisz A, Tousseyn T, Rijal Upadhaya A, Balakrishnan K, Vandenberghe R, Vandenbulcke M, von Arnim CAF, Otto M, Beach TG, Lilja J, Heurling K, Chakrabarty A, Ismail A, Buckley C, Smith APL, Kumar S, Farrar G, Walter J (2019) Different aspects of Alzheimer's disease-related amyloid beta-peptide pathology and their relationship to amyloid positron emission tomography imaging and dementia. Acta Neuropathol Commun 7:178 82. Mattsson N, Palmqvist S, Stomrud E, Vogel J, Hansson O (2019) Staging beta-amyloid pathology with amyloid positron emission tomography. JAMA Neurol 76:1319-1329 83. Ikonomovic MD, Buckley CJ, Abrahamson EE, Kofler JK, Mathis CA, Klunk WE, Farrar G (2020) Post-mortem analyses of PiB and flutemetamol in diffuse and cored amyloid-beta plaques in Alzheimer's disease. Acta Neuropathol 140:463-476 84. Attems J (2005) Sporadic cerebral amyloid angiopathy: pathology, clinical implications, and possible pathomechanisms. Acta Neuropathol 110:345-359 85. Attems J, Jellinger K, Thal DR, Van Nostrand W (2011) Review: sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 37:75-93 86. Thal DR, Griffin WS, de Vos RA, Ghebremedhin E (2008) Cerebral amyloid angiopathy and its relationship to Alzheimer's disease. Acta Neuropathol 115:599-609 87. Allen N, Robinson AC, Snowden J, Davidson YS, Mann DM (2014) Patterns of cerebral amyloid angiopathy define histopathological phenotypes in Alzheimer's disease. Neuropathol Appl Neurobiol 40:136-148 88. Love S, Chalmers K, Ince P, Esiri M, Attems J, Jellinger K, Yamada M, McCarron M, Minett T, Matthews F, Greenberg S, Mann D, Kehoe PG (2014) Development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post-mortem brain tissue. Am J Neurodegener Dis 3:19-32 89. Thal DR, Ghebremedhin E, Rub U, Yamaguchi H, Del Tredici K, Braak H (2002) Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol 61:282-293 90. Nortley R, Korte N, Izquierdo P, Hirunpattarasilp C, Mishra A, Jaunmuktane Z, Kyrargyri V, Pfeiffer T, Khennouf L, Madry C, Gong H, Richard-Loendt A, Huang W, Saito T, Saido TC, Brandner S, Sethi H, Attwell D (2019) Amyloid beta oligomers constrict human capillaries in Alzheimer's disease via signaling to pericytes. Science 365:eaav9518 doi 9510.1126/science.aav9518 91. Attems J, Jellinger KA (2014) Pathologic aspects of the hemorrhagic consequences of small vessel disease on the brain. In: Pantoni L, Gorelick PB (eds) Cerebral Small Vessel Disease. Cambridge University Press Cambridge, UK, pp 29-41 92. Planton M, Pariente J, Nemmi F, Albucher JF, Calviere L, Viguier A, Olivot JM, Salabert AS, Payoux P, Peran P, Raposo N (2020) Interhemispheric distribution of amyloid and small vessel disease burden in cerebral amyloid angiopathy-related intracerebral hemorrhage. Eur J Neurol 27:1664-1671 93. Schreiber S, Wilisch-Neumann A, Schreiber F, Assmann A, Scheumann V, Perosa V, Jandke S, Mawrin C, Carare RO, Werring DJ (2020) The spectrum of age-related small vessel diseases: potential overlap and interactions of amyloid and nonamyloid vasculopathies. Neuropathol Appl Neurobiol 46:219-239 94. Lafon PA, Wang Y, Arango-Lievano M, Torrent J, Salvador-Prince L, Mansuy M, Mestre-Francès N, Givalois L, Liu J, Mercader JV, Jeanneteau F, Desrumaux C, Perrier V (2020) Fungicide residues exposure and beta-amyloid aggregation in a mouse model of Alzheimer's disease. Environ Health Perspect 128:17011 95. Gatti L, Tinelli F, Scelzo E, Arioli F, Di Fede G, Obici L, Pantoni L, Giaccone G, Caroppo P, Parati EA, Bersano A (2020) Understanding the pathophysiology of cerebral amyloid angiopathy. Int J Mol Sci 21 96. Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, van Veluw SJ (2020) Cerebral amyloid angiopathy and Alzheimer disease - one peptide, two pathways. Nat Rev Neurol 16:30-42 97. Revesz T, Ghiso J, Lashley T, Plant G, Rostagno A, Frangione B, Holton JL (2003) Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view. J Neuropathol Exp Neurol 62:885-898 98. Neve RL, Harris P, Kosik KS, Kurnit DM, Donlon TA (1986) Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res 387:271-280 99. Guo T, Noble W, Hanger DP (2017) Roles of tau protein in health and disease. Acta Neuropathol 133:665-704 100. Holmes BB, Diamond MI (2014) Prion-like properties of Tau protein: the importance of extracellular Tau as a therapeutic target. J Biol Chem 289:19855-19861 101. Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI (2012) Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem 287:19440-19451 102. de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, Hyman BT (2010) Caspase activation precedes and leads to tangles. Nature 464:1201-1204 103. Younas N, Zafar S, Shafiq M, Noor A, Siegert A, Arora AS, Galkin A, Zafar A, Schmitz M, Stadelmann C, Andreoletti O, Ferrer I, Zerr I (2020) SFPQ and tau: critical factors contributing to rapid progression of Alzheimer's disease. Acta Neuropathol 140:317-339 104. Mamun AA, Uddin MS, Mathew B, Ashraf GM (2020) Toxic tau: structural origins of tau aggregation in Alzheimer's disease. Neural Regen Res 15:1417-1420 105. Ballatore C, Lee VM, Trojanowski JQ (2007) Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci 8:663-672 106. Guha S, Johnson GVW, Nehrke K (2020) The crosstalk between pathological tau phosphorylation and mitochondrial dysfunction as a key to understanding and treating Alzheimer's disease. Mol Neurobiol 57:5103-5120 107. Xia Y, Prokop S, Gorion KM, Kim JD, Sorrentino ZA, Bell BM, Manaois AN, Chakrabarty P, Davies P, Giasson BI (2020) Tau Ser208 phosphorylation promotes aggregation and reveals neuropathologic diversity in Alzheimer's disease and other tauopathies. Acta Neuropathol Commun 8:88 108. Wischik CM, Novak M, Thogersen HC, Edwards PC, Runswick MJ, Jakes R, Walker JE, Milstein C, Roth M, Klug A (1988) Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A 85:4506-4510 109. Uchihara T (2020) Neurofibrillary changes undergoing morphological and biochemical changes - How does tau with the profile shift of from four repeat to three repeat spread in Alzheimer brain? Neuropathology online Jul 22: doi: 10.1111/neup.12669 110. Kidd M (1964) Alzheimer's disease - an electron microscopical study. Brain 87:307-320 111. Pollanen MS, Markiewicz P, Bergeron C, Goh MC (1994) Twisted ribbon structure of paired helical filaments revealed by atomic force microscopy. Am J Pathol 144:869-873 112. Crowther RA (1991) Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc Natl Acad Sci U S A 88:2288-2292 113. Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, Scheres SHW (2017) Cryo-EM structures of tau filaments from Alzheimer's disease. Nature 547:185-190 114. Falcon B, Zhang W, Schweighauser M, Murzin AG, Vidal R, Garringer HJ, Ghetti B, Scheres SHW, Goedert M (2018) Tau filaments from multiple cases of sporadic and inherited Alzheimer's disease adopt a common fold. Acta Neuropathol 136:699-708 115. Braak E, Braak H, Mandelkow EM (1994) A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol 87:554-567 116. Streit WJ, Khoshbouei H, Bechmann I (2020) Dystrophic microglia in late-onset Alzheimer's disease. Glia 68:845-854 117. Malpas CB, Sharmin S, Kalincik T (2020) The histopathological staging of tau, but not amyloid, corresponds to antemortem cognitive status, dementia stage, functional abilities and neuropsychiatric symptoms. Int J Neurosci Apr 30: doi: 10.1080/00207454.00202020.01758087 118. Andrade-Moraes CH, Oliveira-Pinto AV, Castro-Fonseca E, da Silva CG, Guimaraes DM, Szczupak D, Parente-Bruno DR, Carvalho LR, Polichiso L, Gomes BV, Oliveira LM, Rodriguez RD, Leite RE, Ferretti-Rebustini RE, Jacob-Filho W, Pasqualucci CA, Grinberg LT, Lent R (2013) Cell number changes in Alzheimer's disease relate to dementia, not to plaques and tangles. Brain 136:3738-3752 119. Kuchibhotla KV, Wegmann S, Kopeikina KJ, Hawkes J, Rudinskiy N, Andermann ML, Spires-Jones TL, Bacskai BJ, Hyman BT (2014) Neurofibrillary tangle-bearing neurons are functionally integrated in cortical circuits in vivo. Proc Natl Acad Sci U S A 111:510-514 120. Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309:476-481 121. Spires TL, Orne JD, SantaCruz K, Pitstick R, Carlson GA, Ashe KH, Hyman BT (2006) Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am J Pathol 168:1598-1607 122. Drummond E, Pires G, MacMurray C, Askenazi M, Nayak S, Bourdon M, Safar J, Ueberheide B, Wisniewski T (2020) Phosphorylated tau interactome in the human Alzheimer's disease brain. Brain 143:2803-2817 123. Braak H, Del Tredici K (2011) Alzheimer's pathogenesis: is there neuron-to-neuron propagation? Acta Neuropathol 121:589-595 124. Clavaguera F, Duyckaerts C, Haik S (2020) Prion-like properties of tau assemblies. Curr Opin Neurobiol 61:49-57 125. Colin M, Dujardin S, Schraen-Maschke S, Meno-Tetang G, Duyckaerts C, Courade JP, Buee L (2020) From the prion-like propagation hypothesis to therapeutic strategies of anti-tau immunotherapy. Acta Neuropathol 139:3-25 126. DeVos SL, Corjuc BT, Oakley DH, Nobuhara CK, Bannon RN, Chase A, Commins C, Gonzalez JA, Dooley PM, Frosch MP, Hyman BT (2018) Synaptic tau seeding precedes tau pathology in human Alzheimer's disease brain. Front Neurosci 12:267 127. Dujardin S, Hyman BT (2019) Tau prion-like propagation: state of the art and current challenges. Adv Exp Med Biol 1184:305-325 128. Stancu IC, Vasconcelos B, Ris L, Wang P, Villers A, Peeraer E, Buist A, Terwel D, Baatsen P, Oyelami T, Pierrot N, Casteels C, Bormans G, Kienlen-Campard P, Octave JN, Moechars D, Dewachter I (2015) Templated misfolding of tau by prion-like seeding along neuronal connections impairs neuronal network function and associated behavioral outcomes in tau transgenic mice. Acta Neuropathol 129:875-894 129. Narasimhan S, Guo JL, Changolkar L, Stieber A, McBride JD, Silva LV, He Z, Zhang B, Gathagan RJ, Trojanowski JQ, Lee VMY (2017) Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J Neurosci 37:11406-11423 130. Hopp SC, Lin Y, Oakley D, Roe AD, DeVos SL, Hanlon D, Hyman BT (2018) The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer's disease. J Neuroinflammation 15:269 131. Furman JL, Vaquer-Alicea J, White CL, 3rd, Cairns NJ, Nelson PT, Diamond MI (2017) Widespread tau seeding activity at early Braak stages. Acta Neuropathol 133:91-100 132. Jagust WJ (2020) Imaging tau pathology - the next step. JAMA Neurol 77:796-797 133. Lowe VJ, Lundt ES, Albertson SM, Przybelski SA, Senjem ML, Parisi JE, Kantarci K, Boeve B, Jones DT, Knopman D, Jack CR, Jr., Dickson DW, Petersen RC, Murray ME (2019) Neuroimaging correlates with neuropathologic schemes in neurodegenerative disease. Alzheimers Dement 15:927-939 134. Marquié M, Siao Tick Chong M, Antón-Fernández A, Verwer EE, Sáez-Calveras N, Meltzer AC, Ramanan P, Amaral AC, Gonzalez J, Normandin MD, Frosch MP, Gómez-Isla T (2017) [F-18]-AV-1451 binding correlates with postmortem neurofibrillary tangle Braak staging. Acta Neuropathol 134:619-628 135. Ossenkoppele R, Schonhaut DR, Schöll M, Lockhart SN, Ayakta N, Baker SL, O'Neil JP, Janabi M, Lazaris A, Cantwell A, Vogel J, Santos M, Miller ZA, Bettcher BM, Vossel KA, Kramer JH, Gorno-Tempini ML, Miller BL, Jagust WJ, Rabinovici GD (2016) Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain 139:1551-1567 136. Schwarz AJ, Yu P, Miller BB, Shcherbinin S, Dickson J, Navitsky M, Joshi AD, Devous MD, Sr., Mintun MS (2016) Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain 139:1539-1550 137. Vogels T, Leuzy A, Cicognola C, Ashton NJ, Smolek T, Novak M, Blennow K, Zetterberg H, Hromadka T, Zilka N, Schöll M (2020) Propagation of tau pathology: Integrating insights from postmortem and in vivo studies. Biol Psychiatry 87:808-818 138. Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 42:631-639 139. Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Del Tredici K, Duyckaerts C, Frosch MP, Haroutunian V, Hof PR, Hulette CM, Hyman BT, Iwatsubo T, Jellinger KA, Jicha GA, Kovari E, Kukull WA, Leverenz JB, Love S, Mackenzie IR, Mann DM, Masliah E, McKee AC, Montine TJ, Morris JC, Schneider JA, Sonnen JA, Thal DR, Trojanowski JQ, Troncoso JC, Wisniewski T, Woltjer RL, Beach TG (2012) Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol 71:362-381 140. Ohm DT, Fought AJ, Rademaker A, Kim G, Sridhar J, Coventry C, Gefen T, Weintraub S, Bigio E, Mesulam MM, Rogalski E, Geula C (2020) Neuropathologic basis of in vivo cortical atrophy in the aphasic variant of Alzheimer's disease. Brain Pathol 30:332-344 141. Kaufman SK, Del Tredici K, Thomas TL, Braak H, Diamond MI (2018) Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer's disease and PART. Acta Neuropathol 136:57-67 142. Attems J, Thomas A, Jellinger K (2012) Correlations between cortical and subcortical tau pathology. Neuropathol Appl Neurobiol 38:582-590 143. Braak H, Thal DR, Ghebremedhin E, Del Tredici K (2011) Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 70:960-969 144. Grinberg LT, Rüb U, Ferretti RE, Nitrini R, Farfel JM, Polichiso L, Gierga K, Jacob-Filho W, Heinsen H (2009) The dorsal raphe nucleus shows phospho-tau neurofibrillary changes before the transentorhinal region in Alzheimer's disease. A precocious onset? Neuropathol Appl Neurobiol 35:406-416 145. Braak H, Del Trecidi K (2015) Neuroanatomy and pathology of sporadic Alzheimer's disease. Adv Anat Embryol Cell Biol 215:1-162 146. Grudzien A, Shaw P, Weintraub S, Bigio E, Mash DC, Mesulam MM (2007) Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer's disease. Neurobiol Aging 28:327-335 147. Rüb U, Stratmann K, Heinsen H, Turco DD, Seidel K, Dunnen W, Korf HW (2016) The brainstem tau cytoskeletal pathology of Alzheimer's disease: a brief historical overview and description of its anatomical distribution pattern, evolutional features, pathogenetic and clinical relevance. Curr Alzheimer Res 13:1178-1197 148. Attems J, Quass M, Jellinger KA (2007) Tau and alpha-synuclein brainstem pathology in Alzheimer disease: relation with extrapyramidal signs. Acta Neuropathol 113:53-62 149. Buchman AS, Shulman JM, Nag S, Leurgans SE, Arnold SE, Morris MC, Schneider JA, Bennett DA (2012) Nigral pathology and parkinsonian signs in elders without Parkinson disease. Ann Neurol 71:258-266 150. Burns JM, Galvin JE, Roe CM, Morris JC, McKeel DW (2005) The pathology of the substantia nigra in Alzheimer disease with extrapyramidal signs. Neurology 64:1397-1403 151. Kent SA, Spires-Jones TL, Durrant CS (2020) The physiological roles of tau and Abeta: implications for Alzheimer's disease pathology and therapeutics. Acta Neuropathol 140:417-447 152. Kara E, Marks JD, Aguzzi A (2018) Toxic protein spread in neurodegeneration: reality versus fantasy. Trends Mol Med 24:1007-1020 153. Busche MA, Hyman BT (2020) Synergy between amyloid-beta and tau in Alzheimer's disease. Nat Neurosci 23:1183-1193 154. Jucker M, Walker LC (2018) Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat Neurosci 21:1341-1349 155. Thompson TB, Chaggar P, Kuhl E, Goriely A (2020) Protein-protein interactions in neurodegenerative diseases: A conspiracy theory. PLoS Comput Biol 16:e1008267 156. Bennett RE, DeVos SL, Dujardin S, Corjuc B, Gor R, Gonzalez J, Roe AD, Frosch MP, Pitstick R, Carlson GA, Hyman BT (2017) Enhanced tau aggregation in the presence of amyloid beta. Am J Pathol 187:1601-1612 157. Pooler AM, Polydoro M, Maury EA, Nicholls SB, Reddy SM, Wegmann S, William C, Saqran L, Cagsal-Getkin O, Pitstick R, Beier DR, Carlson GA, Spires-Jones TL, Hyman BT (2015) Amyloid accelerates tau propagation and toxicity in a model of early Alzheimer's disease. Acta Neuropathol Commun 3:14 158. Cline EN, Bicca MA, Viola KL, Klein WL (2018) The amyloid-ß oligomer hypothesis: beginning of the third decade. J Alzheimers Dis 64:S567-S610 159. Iadanza MG, Jackson MP, Hewitt EW, Ranson NA, Radford SE (2018) A new era for understanding amyloid structures and disease. Nat Rev Mol Cell Biol 19:755-773 160. Okamura N, Harada R, Furumoto S, Arai H, Yanai K, Kudo Y (2014) Tau PET imaging in Alzheimer's disease. Curr Neurol Neurosci Rep 14:500 161. Keene CD, Wilson AM, Kilgore MD, Bruner LT, Postupna NO, Darvas M (2019) Luminex-based quantification of Alzheimer's disease neuropathologic change in formalin-fixed post-mortem human brain tissue. Lab Invest 99:1056-1067 162. Walsh DM, Selkoe DJ (2020) Amyloid beta-protein and beyond: the path forward in Alzheimer's disease. Curr Opin Neurobiol 61:116-124 163. Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med 8:595-608 164. Viola KL, Klein WL (2015) Amyloid beta oligomers in Alzheimer's disease pathogenesis, treatment, and diagnosis. Acta Neuropathol 129:183-206 165. Gomes LA, Hipp SA, Rijal Upadhaya A, Balakrishnan K, Ospitalieri S, Koper MJ, Largo-Barrientos P, Uytterhoeven V, Reichwald J, Rabe S, Vandenberghe R, von Arnim CAF, Tousseyn T, Feederle R, Giudici C, Willem M, Staufenbiel M, Thal DR (2019) Abeta-induced acceleration of Alzheimer-related tau-pathology spreading and its association with prion protein. Acta Neuropathol 138:913-941 166. Vergara C, Houben S, Suain V, Yilmaz Z, De Decker R, Vanden Dries V, Boom A, Mansour S, Leroy K, Ando K, Brion JP (2019) Amyloid-beta pathology enhances pathological fibrillary tau seeding induced by Alzheimer PHF in vivo. Acta Neuropathol 137:397-412 167. Wu HY, Kuo PC, Wang YT, Lin HT, Roe AD, Wang BY, Han CL, Hyman BT, Chen YJ, Tai HC (2018) Beta-amyloid induces pathology-related patterns of tau hyperphosphorylation at synaptic terminals. J Neuropathol Exp Neurol 77:814-826 168. He Z, Guo JL, McBride JD, Narasimhan S, Kim H, Changolkar L, Zhang B, Gathagan RJ, Yue C, Dengler C, Stieber A, Nitla M, Coulter DA, Abel T, Brunden KR, Trojanowski JQ, Lee VM (2018) Amyloid-ß plaques enhance Alzheimer's brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat Med 24:29-38 169. De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, Bigio EH, Jerecic J, Acton PJ, Shughrue PJ, Chen-Dodson E, Kinney GG, Klein WL (2008) Alzheimer's disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging 29:1334-1347 170. Shin WS, Di J, Cao Q, Li B, Seidler PM, Murray KA, Bitan G, Jiang L (2019) Amyloid beta-protein oligomers promote the uptake of tau fibril seeds potentiating intracellular tau aggregation. Alzheimers Res Ther 11:86 171. Welikovitch LA, Do Carmo S, Maglóczky Z, Szocsics P, Loke J, Freund T, Cuello AC (2018) Evidence of intraneuronal Abeta accumulation preceding tau pathology in the entorhinal cortex. Acta Neuropathol 136:901-917 172. Wang L, Benzinger TL, Su Y, Christensen J, Friedrichsen K, Aldea P, McConathy J, Cairns NJ, Fagan AM, Morris JC, Ances BM (2016) Evaluation of tau imaging in staging Alzheimer disease and revealing interactions between beta-amyloid and tauopathy. JAMA Neurol 73:1070-1077 173. Sardar Sinha M, Ansell-Schultz A, Civitelli L, Hildesjö C, Larsson M, Lannfelt L, Ingelsson M, Hallbeck M (2018) Alzheimer's disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol 136:41-56 174. King A, Bodi I, Troakes C (2020) The neuropathological diagnosis of Alzheimer's disease - the challenges of pathological mimics and concomitant pathology. Brain Sci 10:479 175. Gulisano W, Maugeri D, Baltrons MA, Fà M, Amato A, Palmeri A, D'Adamio L, Grassi C, Devanand DP, Honig LS, Puzzo D, Arancio O (2018) Role of amyloid-beta and tau proteins in Alzheimer's disease: confuting the amyloid cascade. J Alzheimers Dis 64:S611-S631 176. Small SA, Duff K (2008) Linking Abeta and tau in late-onset Alzheimer's disease: a dual pathway hypothesis. Neuron 60:534-542 177. Rice HC, de Malmazet D, Schreurs A, Frere S, Van Molle I, Volkov AN, Creemers E, Vertkin I, Nys J, Ranaivoson FM, Comoletti D, Savas JN, Remaut H, Balschun D, Wierda KD, Slutsky I, Farrow K, De Strooper B, de Wit J (2019) Secreted amyloid-beta precursor protein functions as a GABABR1a ligand to modulate synaptic transmission. Science 363:eaao4827, doi 4810.1126/science.aao4827 178. Ruiz-Riquelme A, Lau HHC, Stuart E, Goczi AN, Wang Z, Schmitt-Ulms G, Watts JC (2018) Prion-like propagation of beta-amyloid aggregates in the absence of APP overexpression. Acta Neuropathol Commun 6:26 179. Pickett EK, Koffie RM, Wegmann S, Henstridge CM, Herrmann AG, Colom-Cadena M, Lleo A, Kay KR, Vaught M, Soberman R, Walsh DM, Hyman BT, Spires-Jones TL (2016) Non-fibrillar oligomeric amyloid-beta within synapses. J Alzheimers Dis 53:787-800 180. Mroczko B, Groblewska M, Litman-Zawadzka A, Kornhuber J, Lewczuk P (2018) Amyloid ß oligomers (AßOs) in Alzheimer's disease. J Neural Transm (Vienna) 125:177-191 181. Koss DJ, Jones G, Cranston A, Gardner H, Kanaan NM, Platt B (2016) Soluble pre-fibrillar tau and beta-amyloid species emerge in early human Alzheimer's disease and track disease progression and cognitive decline. Acta Neuropathol 132:875-895 182. Takahashi RH, Capetillo-Zarate E, Lin MT, Milner TA, Gouras GK (2010) Co-occurrence of Alzheimer's disease beta-amyloid and tau pathologies at synapses. Neurobiol Aging 31:1145-1152 183. Rajmohan R, Reddy PH (2017) Amyloid-beta and phosphorylated tau accumulations cause abnormalities at synapses of Alzheimer's disease neurons. J Alzheimers Dis 57:975-999 184. van der Kant R, Goldstein LSB, Ossenkoppele R (2020) Amyloid-beta-independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci 21:21-35 185. Horie K, Barthélemy NR, Mallipeddi N, Li Y, Franklin EE, Perrin RJ, Bateman RJ, Sato C (2020) Regional correlation of biochemical measures of amyloid and tau phosphorylation in the brain. Acta Neuropathol Commun 8:149 186. Tripathi T, Khan H (2020) Direct interaction between the beta-amyloid core and tau facilitates cross-seeding: a novel target for therapeutic intervention. Biochemistry 59:341-342 187. Catania M, Di Fede G (2020) One or more beta-amyloid(s)? New insights into the prion-like nature of Alzheimer's disease. Prog Mol Biol Transl Sci 175:213-237 188. Panza F, Lozupone M, Dibello V, Greco A, Daniele A, Seripa D, Logroscino G, Imbimbo BP (2019) Are antibodies directed against amyloid-beta (Abeta) oligomers the last call for the Abeta hypothesis of Alzheimer's disease? Immunotherapy 11:3-6 189. Kuang H, Tan CY, Tian HZ, Liu LH, Yang MW, Hong FF, Yang SL (2020) Exploring the bi-directional relationship between autophagy and Alzheimer's disease. CNS Neurosci Ther 26:155-166 190. Jaunmuktane Z, Brandner S (2020) The role of prion-like mechanisms in neurodegenerative diseases. Neuropathol Appl Neurobiol 46:522-545 191. Penke B, Szucs M, Bogár F (2020) Oligomerization and conformational change turn monomeric beta-amyloid and tau proteins toxic: their role in Alzheimer's pathogenesis. Molecules 25 192. Dujardin S, Commins C, Lathuiliere A, Beerepoot P, Fernandes AR, Kamath TV, De Los Santos MB, Klickstein N, Corjuc DL, Corjuc BT, Dooley PM, Viode A, Oakley DH, Moore BD, Mullin K, Jean-Gilles D, Clark R, Atchison K, Moore R, Chibnik LB, Tanzi RE, Frosch MP, Serrano-Pozo A, Elwood F, Steen JA, Kennedy ME, Hyman BT (2020) Tau molecular diversity contributes to clinical heterogeneity in Alzheimer's disease. Nat Med 26:1256-1263 193. Sintini I, Graff-Radford J, Senjem ML, Schwarz CG, Machulda MM, Martin PR, Jones DT, Boeve BF, Knopman DS, Kantarci K, Petersen RC, Jack CR, Lowe VJ, Josephs KA, Whitwell JL (2020) Longitudinal neuroimaging biomarkers differ across Alzheimer's disease phenotypes. Brain 143:2281-2294 194. Pascoal TA, Mathotaarachchi S, Shin M, Benedet AL, Mohades S, Wang S, Beaudry T, Kang MS, Soucy JP, Labbe A, Gauthier S, Rosa-Neto P (2017) Synergistic interaction between amyloid and tau predicts the progression to dementia. Alzheimers Dement 13:644-653 195. Thal DR, Griffin WS, Braak H (2008) Parenchymal and vascular Abeta-deposition and its effects on the degeneration of neurons and cognition in Alzheimer's disease. J Cell Mol Med 12:1848-1862 196. Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT (1997) Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol 41:17-24 197. Saez-Atienzar S, Masliah E (2020) Cellular senescence and Alzheimer disease: the egg and the chicken scenario. Nat Rev Neurosci 21:433-444 198. Brewer GJ, Herrera RA, Philipp S, Sosna J, Reyes-Ruiz JM, Glabe CG (2020) Age-related intraneuronal aggregation of amyloid-beta in endosomes, mitochondria, autophagosomes, and lysosomes. J Alzheimers Dis 73:229-246 199. Llorian M, Schwartz S, Clark TA, Hollander D, Tan LY, Spellman R, Gordon A, Schweitzer AC, de la Grange P, Ast G, Smith CW (2010) Position-dependent alternative splicing activity revealed by global profiling of alternative splicing events regulated by PTB. Nat Struct Mol Biol 17:1114-1123 200. Zanjani H, Finch CE, Kemper C, Atkinson J, McKeel D, Morris JC, Price JL (2005) Complement activation in very early Alzheimer disease. Alzheimer Dis Assoc Disord 19:55-66 201. Kordower JH, Chu Y, Stebbins GT, DeKosky ST, Cochran EJ, Bennett D, Mufson EJ (2001) Loss and atrophy of layer II entorhinal cortex neurons in elderly people with mild cognitive impairment. Ann Neurol 49:202-213 202. Duyckaerts C (2014) Alzheimer’s disease. In: Kovacs GG (ed) Neuropathology of Neurodegenerative Diseases. A Practical Guide. Cambridge University Press Cambridge, pp 80-108 203. Jellinger KA (2006) Challenges in neuronal apoptosis. Curr Alzheimer Res 3:377-391 204. Woodhouse A, Dickson TC, West AK, McLean CA, Vickers JC (2006) No difference in expression of apoptosis-related proteins and apoptotic morphology in control, pathologically aged and Alzheimer's disease cases. Neurobiol Dis 22:323-333 205. Gorlovoy P, Larionov S, Pham TT, Neumann H (2009) Accumulation of tau induced in neurites by microglial proinflammatory mediators. FASEB J 23:2502-2513 206. Li JW, Zong Y, Cao XP, Tan L (2018) Microglial priming in Alzheimer's disease. Ann Transl Med 6:176 207. Forner S, Baglietto-Vargas D, Martini AC, Trujillo-Estrada L, LaFerla FM (2017) Synaptic impairment in Alzheimer's disease: a dysregulated symphony. Trends Neurosci 40:347-357 208. Scheff SW, Price DA (2003) Synaptic pathology in Alzheimer's disease: a review of ultrastructural studies. Neurobiol Aging 24:1029-1046 209. Scheff SW, Price DA, Schmitt FA, Mufson EJ (2006) Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiol Aging 27:1372-1384 210. Smith HL, Freeman OJ, Butcher AJ, Holmqvist S, Humoud I, Schätzl T, Hughes DT, Verity NC, Swinden DP, Hayes J, de Weerd L, Rowitch DH, Franklin RJM, Mallucci GR (2020) Astrocyte unfolded protein response induces a specific reactivity state that causes non-cell-autonomous neuronal degeneration. Neuron 105:855-866 e855 211. Elliott C, Rojo AI, Ribe E, Broadstock M, Xia W, Morin P, Semenov M, Baillie G, Cuadrado A, Al-Shawi R, Ballard CG, Simons P, Killick R (2018) A role for APP in Wnt signalling links synapse loss with beta-amyloid production. Transl Psychiatry 8:179 212. Marshall GA, Fairbanks LA, Tekin S, Vinters HV, Cummings JL (2007) Early-onset Alzheimer's disease is associated with greater pathologic burden. J Geriatr Psychiatry Neurol 20:29-33 213. Scheff SW, Neltner JH, Nelson PT (2014) Is synaptic loss a unique hallmark of Alzheimer's disease? Biochem Pharmacol 88:517-528 214. Yin Z, Raj D, Saiepour N, Van Dam D, Brouwer N, Holtman IR, Eggen BJL, Möller T, Tamm JA, Abdourahman A, Hol EM, Kamphuis W, Bayer TA, De Deyn PP, Boddeke E (2017) Immune hyperreactivity of Abeta plaque-associated microglia in Alzheimer's disease. Neurobiol Aging 55:115-122 215. Zotova E, Bharambe V, Cheaveau M, Morgan W, Holmes C, Harris S, Neal JW, Love S, Nicoll JA, Boche D (2013) Inflammatory components in human Alzheimer's disease and after active amyloid-beta42 immunization. Brain 136:2677-2696 216. Britschgi M, Takeda-Uchimura Y, Rockenstein E, Johns H, Masliah E, Wyss-Coray T (2012) Deficiency of terminal complement pathway inhibitor promotes neuronal tau pathology and degeneration in mice. J Neuroinflammation 9:220 217. Bolós M, Llorens-Martín M, Jurado-Arjona J, Hernández F, Rábano A, Avila J (2016) Direct evidence of internalization of tau by microglia in vitro and in vivo. J Alzheimers Dis 50:77-87 218. Luo W, Liu W, Hu X, Hanna M, Caravaca A, Paul SM (2015) Microglial internalization and degradation of pathological tau is enhanced by an anti-tau monoclonal antibody. Sci Rep 5:11161 219. van Olst L, Verhaege D, Franssen M, Kamermans A, Roucourt B, Carmans S, Ytebrouck E, van der Pol SMA, Wever D, Popovic M, Vandenbroucke RE, Sobrino T, Schouten M, de Vries HE (2020) Microglial activation arises after aggregation of phosphorylated-tau in a neuron-specific P301S tauopathy mouse model. Neurobiol Aging 89:89-98 220. Zhu K, Pieber M, Han J, Blomgren K, Zhang XM, Harris RA, Lund H (2020) Absence of microglia or presence of peripherally-derived macrophages does not affect tau pathology in young or old hTau mice. Glia 68:1466-1478 221. Walker DG (2020) Defining activation states of microglia in human brain tissue: an unresolved issue for Alzheimer’s disease. Neuroimmunol Neuroinflammation 7:194-214; doi: 110.20517/22347-28659.22020.20509 222. Paasila PJ, Davies DS, Kril JJ, Goldsbury C, Sutherland GT (2019) The relationship between the morphological subtypes of microglia and Alzheimer's disease neuropathology. Brain Pathol 29:726-740 223. Baulch JE, Acharya MM, Agrawal S, Apodaca LA, Monteiro C, Agrawal A (2020) Immune and inflammatory determinants underlying Alzheimer's disease pathology. J Neuroimmune Pharmacol Feb 22: doi: 10.1007/s11481-11020-09908-11489 224. Jicha GA, Parisi JE, Dickson DW, Johnson K, Cha R, Ivnik RJ, Tangalos EG, Boeve BF, Knopman DS, Braak H, Petersen RC (2006) Neuropathologic outcome of mild cognitive impairment following progression to clinical dementia. Arch Neurol 63:674-681 225. Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Jr., Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH (2011) Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 7:280-292 226. Thal DR, von Arnim C, Griffin WS, Yamaguchi H, Mrak RE, Attems J, Upadhaya AR (2013) Pathology of clinical and preclinical Alzheimer's disease. Eur Arch Psychiatry Clin Neurosci 263 Suppl 2:S137-145 227. Jack CR, Wiste HJ, Weigand SD, Therneau TM, Lowe VJ, Knopman DS, Botha H, Graff-Radford J, Jones DT, Ferman TJ, Boeve BF, Kantarci K, Vemuri P, Mielke MM, Whitwell J, Josephs K, Schwarz CG, Senjem ML, Gunter JL, Petersen RC (2020) Predicting future rates of tau accumulation on PET. Brain 143:3136-3150 228. Alafuzoff I, Arzberger T, Al-Sarraj S, Bodi I, Bogdanovic N, Braak H, Bugiani O, Del-Tredici K, Ferrer I, Gelpi E, Giaccone G, Graeber MB, Ince P, Kamphorst W, King A, Korkolopoulou P, Kovacs GG, Larionov S, Meyronet D, Monoranu C, Parchi P, Patsouris E, Roggendorf W, Seilhean D, Tagliavini F, Stadelmann C, Streichenberger N, Thal DR, Wharton SB, Kretzschmar H (2008) Staging of neurofibrillary pathology in Alzheimer's disease: a study of the BrainNet Europe Consortium. Brain Pathol 18:484-496 229. Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ (2012) National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement 8:1-13 230. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L (1991) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41:479-486 231. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112:389-404 232. Jellinger KA (2009) Criteria for the neuropathological diagnosis of dementing disorders: routes out of the swamp? Acta Neuropathol 117:101-110 233. Jellinger KA (2009) A critical evaluation of current staging of alpha-synuclein pathology in Lewy body disorders. Biochim Biophys Acta 1792:730-740 234. Hyman BT, Trojanowski JQ (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol 56:1095-1097 235. Bowler JV, Munoz DG, Merskey H, Hachinski V (1998) Fallacies in the pathological confirmation of the diagnosis of Alzheimer's disease. J Neurol Neurosurg Psychiatry 64:18-24. 236. Beach TG, Monsell SE, Phillips LE, Kukull W (2012) Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005-2010. J Neuropathol Exp Neurol 71:266-273 237. Montine TJ, Monsell SE, Beach TG, Bigio EH, Bu Y, Cairns NJ, Frosch M, Henriksen J, Kofler J, Kukull WA, Lee EB, Nelson PT, Schantz AM, Schneider JA, Sonnen JA, Trojanowski JQ, Vinters HV, Zhou XH, Hyman BT (2016) Multisite assessment of NIA-AA guidelines for the neuropathologic evaluation of Alzheimer's disease. Alzheimers Dement 12:164-169 238. Plassman BL, Khachaturian AS, Townsend JJ, Ball MJ, Steffens DC, Leslie CE, Tschanz JT, Norton MC, Burke JR, Welsh-Bohmer KA, Hulette CM, Nixon RR, Tyrey M, Breitner JC (2006) Comparison of clinical and neuropathologic diagnoses of Alzheimer's disease in 3 epidemiologic samples. Alzheimers Dement 2:2-11 239. Cure S, Abrams K, Belger M, Dell'agnello G, Happich M (2014) Systematic literature review and meta-analysis of diagnostic test accuracy in Alzheimer's disease and other dementia using autopsy as standard of truth. J Alzheimers Dis 42:169-182 240. Janocko NJ, Brodersen KA, Soto-Ortolaza AI, Ross OA, Liesinger AM, Duara R, Graff-Radford NR, Dickson DW, Murray ME (2012) Neuropathologically defined subtypes of Alzheimer's disease differ significantly from neurofibrillary tangle-predominant dementia. Acta Neuropathol 124:681-692 241. Jellinger KA (2014) Alzheimer’s disease: current clinical and neuropathologic diagnostic criteria. Austin Alzheimers J Parkinsons Dis 1:6 242. Murray ME, Cannon A, Graff-Radford NR, Liesinger AM, Rutherford NJ, Ross OA, Duara R, Carrasquillo MM, Rademakers R, Dickson DW (2014) Differential clinicopathologic and genetic features of late-onset amnestic dementias. Acta Neuropathol 128:411-421 243. Tiraboschi P, Sabbagh MN, Hansen LA, Salmon DP, Merdes A, Gamst A, Masliah E, Alford M, Thal LJ, Corey-Bloom J (2004) Alzheimer disease without neocortical neurofibrillary tangles: "a second look". Neurology 62:1141-1147 244. Hansen L, Salmon D, Galasko D, Masliah E, Katzman R, DeTeresa R, Thal L, Pay MM, Hofstetter R, Klauber M, et al. (1990) The Lewy body variant of Alzheimer's disease: a clinical and pathologic entity. Neurology 40:1-8. 245. Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, Arnold SE, Attems J, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Gearing M, Grinberg LT, Hof PR, Hyman BT, Jellinger K, Jicha GA, Kovacs GG, Knopman DS, Kofler J, Kukull WA, Mackenzie IR, Masliah E, McKee A, Montine TJ, Murray ME, Neltner JH, Santa-Maria I, Seeley WW, Serrano-Pozo A, Shelanski ML, Stein T, Takao M, Thal DR, Toledo JB, Troncoso JC, Vonsattel JP, White CL, 3rd, Wisniewski T, Woltjer RL, Yamada M, Nelson PT (2014) Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 128:755-766 246. Jellinger KA, Attems J (2007) Neurofibrillary tangle-predominant dementia: comparison with classical Alzheimer disease. Acta Neuropathol 113:107-117 247. Besser LM, Kukull WA, Teylan MA, Bigio EH, Cairns NJ, Kofler JK, Montine TJ, Schneider JA, Nelson PT (2018) The revised National Alzheimer's Coordinating Center's Neuropathology form-available data and new analyses. J Neuropathol Exp Neurol 77:717-726 248. Jellinger KA, Attems J (2015) Challenges of multimorbidity of the aging brain: a critical update. J Neural Transm (Vienna) 122:505-521 249. Josephs KA, Murray ME, Tosakulwong N, Whitwell JL, Knopman DS, Machulda MM, Weigand SD, Boeve BF, Kantarci K, Petrucelli L, Lowe VJ, Jack CR, Jr., Petersen RC, Parisi JE, Dickson DW (2017) Tau aggregation influences cognition and hippocampal atrophy in the absence of beta-amyloid: a clinico-imaging-pathological study of primary age-related tauopathy (PART). Acta Neuropathol 133:705-715 250. Jellinger KA (2018) Different patterns of hippocampal tau pathology in Alzheimer's disease and PART. Acta Neuropathol 136:811-813 251. Zhang L, Jiang Y, Zhu J, Liang H, He X, Qian J, Lin H, Tao Y, Zhu K (2020) Quantitative assessment of hippocampal tau pathology in AD and PART. J Mol Neurosci 70:1808-1811 252. Zhang X, Sun B, Wang X, Lu H, Shao F, Rozemuller AJM, Liang H, Liu C, Chen J, Huang M, Zhu K (2019) Phosphorylated TDP-43 staging of primary age-related tauopathy. Neurosci Bull 35:183-192 253. Jellinger KA, Alafuzoff I, Attems J, Beach TG, Cairns NJ, Crary JF, Dickson DW, Hof PR, Hyman BT, Jack CR, Jr., Jicha GA, Knopman DS, Kovacs GG, Mackenzie IR, Masliah E, Montine TJ, Nelson PT, Schmitt F, Schneider JA, Serrano-Pozo A, Thal DR, Toledo JB, Trojanowski JQ, Troncoso JC, Vonsattel JP, Wisniewski T (2015) PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol 129:757-762 254. Jellinger KA (2016) Commentary on the paper "PART, a Distinct Tauopathy, Different from Classical 1. Sporadic Alzheimer Disease". J Clin Cell Immunol 7:1000480 255. Bancher C, Egensperger R, Kosel S, Jellinger K, Graeber MB (1997) Low prevalence of apolipoprotein E epsilon 4 allele in the neurofibrillary tangle predominant form of senile dementia. Acta Neuropathol 94:403-409 256. McMillan CT, Lee EB, Jefferson-George K, Naj A, Van Deerlin VM, Trojanowski JQ, Wolk DA (2018) Alzheimer's genetic risk is reduced in primary age-related tauopathy: a potential model of resistance? Ann Clin Transl Neurol 5:927-934 257. Hickman RA, Flowers XE, Wisniewski T (2020) Primary age-related tauopathy (PART): addressing the spectrum of neuronal tauopathic changes in the aging brain. Curr Neurol Neurosci Rep 20:39 258. Besser LM, Teylan MA, Nelson PT (2020) Limbic predominant age-related TDP-43 encephalopathy (LATE): clinical and neuropathological associations. J Neuropathol Exp Neurol 79:305-313 259. Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, Rademakers R, Alafuzoff I, Attems J, Brayne C, Coyle-Gilchrist ITS, Chui HC, Fardo DW, Flanagan ME, Halliday G, Hokkanen SRK, Hunter S, Jicha GA, Katsumata Y, Kawas CH, Keene CD, Kovacs GG, Kukull WA, Levey AI, Makkinejad N, Montine TJ, Murayama S, Murray ME, Nag S, Rissman RA, Seeley WW, Sperling RA, White Iii CL, Yu L, Schneider JA (2019) Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 142:1503-1527 260. Robinson JL, Porta S, Garrett FG, Zhang P, Xie SX, Suh E, Van Deerlin VM, Abner EL, Jicha GA, Barber JM, Lee VM, Lee EB, Trojanowski JQ, Nelson PT (2020) Limbic-predominant age-related TDP-43 encephalopathy differs from frontotemporal lobar degeneration. Brain 143:2844-2857 261. Teylan M, Besser LM, Crary JF, Mock C, Gauthreaux K, Thomas NM, Chen YC, Kukull WA (2019) Clinical diagnoses among individuals with primary age-related tauopathy versus Alzheimer's neuropathology. Lab Invest 99:1049-1055 262. Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW (2011) Neuropathologically defined subtypes of Alzheimer's disease with distinct clinical characteristics: a retrospective study. Lancet Neurol 10:785-796 263. Lehmann M, Ghosh PM, Madison C, Laforce R, Jr., Corbetta-Rastelli C, Weiner MW, Greicius MD, Seeley WW, Gorno-Tempini ML, Rosen HJ, Miller BL, Jagust WJ, Rabinovici GD (2013) Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer's disease. Brain 136:844-858 264. Rabinovici GD, Furst AJ, Alkalay A, Racine CA, O'Neil JP, Janabi M, Baker SL, Agarwal N, Bonasera SJ, Mormino EC, Weiner MW, Gorno-Tempini ML, Rosen HJ, Miller BL, Jagust WJ (2010) Increased metabolic vulnerability in early-onset Alzheimer's disease is not related to amyloid burden. Brain 133:512-528 265. Iaccarino L, Tammewar G, Ayakta N, Baker SL, Bejanin A, Boxer AL, Gorno-Tempini ML, Janabi M, Kramer JH, Lazaris A, Lockhart SN, Miller BL, Miller ZA, O'Neil JP, Ossenkoppele R, Rosen HJ, Schonhaut DR, Jagust WJ, Rabinovici GD (2017) Local and distant relationships between amyloid, tau and neurodegeneration in Alzheimer's Disease. Neuroimage Clin 17:452-464 266. Ossenkoppele R, Pijnenburg YA, Perry DC, Cohn-Sheehy BI, Scheltens NM, Vogel JW, Kramer JH, van der Vlies AE, La Joie R, Rosen HJ, van der Flier WM, Grinberg LT, Rozemuller AJ, Huang EJ, van Berckel BN, Miller BL, Barkhof F, Jagust WJ, Scheltens P, Seeley WW, Rabinovici GD (2015) The behavioural/dysexecutive variant of Alzheimer's disease: clinical, neuroimaging and pathological features. Brain 138:2732-2749 267. Tetzloff KA, Graff-Radford J, Martin PR, Tosakulwong N, Machulda MM, Duffy JR, Clark HM, Senjem ML, Schwarz CG, Spychalla AJ, Drubach DA, Jack CR, Lowe VJ, Josephs KA, Whitwell JL (2018) Regional distribution, asymmetry, and clinical correlates of tau uptake on [18F]AV-1451 PET in atypical Alzheimer's disease. J Alzheimers Dis 62:1713-1724 268. Hwang J, Kim CM, Jeon S, Lee JM, Hong YJ, Roh JH, Lee JH, Koh JY, Na DL (2015) Prediction of Alzheimer's disease pathophysiology based on cortical thickness patterns. Alzheimers Dement (Amst) 2:58-67 269. Whitwell JL, Dickson DW, Murray ME, Weigand SD, Tosakulwong N, Senjem ML, Knopman DS, Boeve BF, Parisi JE, Petersen RC, Jack CR, Jr., Josephs KA (2012) Neuroimaging correlates of pathologically defined subtypes of Alzheimer's disease: a case-control study. Lancet Neurol 11:868-877 270. Jellinger KA (2012) Neuropathological subtypes of Alzheimer's disease (Correspondence). Acta Neuropathol 123:153-154 271. McAleese KE, Walker L, Graham S, Moya ELJ, Johnson M, Erskine D, Colloby SJ, Dey M, Martin-Ruiz C, Taylor JP, Thomas AJ, McKeith IG, De Carli C, Attems J (2017) Parietal white matter lesions in Alzheimer's disease are associated with cortical neurodegenerative pathology, but not with small vessel disease. Acta Neuropathol 134:459-473 272. Strain JF, Smith RX, Beaumont H, Roe CM, Gordon BA, Mishra S, Adeyemo B, Christensen JJ, Su Y, Morris JC, Benzinger TLS, Ances BM (2018) Loss of white matter integrity reflects tau accumulation in Alzheimer disease defined regions. Neurology 91:e313-e318 273. Arfanakis K, Evia AM, Leurgans SE, Cardoso LFC, Kulkarni A, Alqam N, Lopes LF, Vieira D, Bennett DA, Schneider JA (2020) Neuropathologic correlates of white matter hyperintensities in a community-based cohort of older adults. J Alzheimers Dis 73:333-345 274. McAleese KE, Colloby S, Attems J, Thomas A, Francis PT (2020) Mixed brain pathologies account for most dementia in the Brains for Dementia Research cohort (abstract). Neuropathol Appl Neurobiol 46 (Suppl. 1):24 275. McAleese KE, Mohi M, Graham S, Baker G, Walker L, DeCarli C, Koss D, Attems J (2020) The aetiology of frontal white matter lesions in Alzheimer’s disease are associated with both neurodegenerative and ischemic mechanisms (abstract). Neuropathol Appl Neurobiol 46 (Suppl. 1):15 276. Ferreira D, Verhagen C, Hernandez-Cabrera JA, Cavallin L, Guo CJ, Ekman U, Muehlboeck JS, Simmons A, Barroso J, Wahlund LO, Westman E (2017) Distinct subtypes of Alzheimer's disease based on patterns of brain atrophy: longitudinal trajectories and clinical applications. Sci Rep 7:46263 277. Ferreira D, Pereira JB, Volpe G, Westman E (2019) Subtypes of Alzheimer's disease display distinct network abnormalities extending beyond their pattern of brain atrophy. Front Neurol 10:524 278. Risacher SL, Anderson WH, Charil A, Castelluccio PF, Shcherbinin S, Saykin AJ, Schwarz AJ (2017) Alzheimer disease brain atrophy subtypes are associated with cognition and rate of decline. Neurology 89:2176-2186 279. Lowe VJ, Lundt ES, Albertson SM, Min HK, Fang P, Przybelski SA, Senjem ML, Schwarz CG, Kantarci K, Boeve B, Jones DT, Reichard RR, Tranovich JF, Hanna Al-Shaikh FS, Knopman DS, Jack CR, Jr., Dickson DW, Petersen RC, Murray ME (2020) Tau-positron emission tomography correlates with neuropathology findings. Alzheimers Dement 16:561-571 280. Galton CJ, Patterson K, Xuereb JH, Hodges JR (2000) Atypical and typical presentations of Alzheimer's disease: a clinical, neuropsychological, neuroimaging and pathological study of 13 cases. Brain 123 Pt 3:484-498 281. Lam B, Masellis M, Freedman M, Stuss DT, Black SE (2013) Clinical, imaging, and pathological heterogeneity of the Alzheimer's disease syndrome. Alzheimers Res Ther 5:1 282. Ahmed S, de Jager CA, Haigh AM, Garrard P (2012) Logopenic aphasia in Alzheimer's disease: clinical variant or clinical feature? J Neurol Neurosurg Psychiatry 83:1056-1062 283. Gorno-Tempini ML, Brambati SM, Ginex V, Ogar J, Dronkers NF, Marcone A, Perani D, Garibotto V, Cappa SF, Miller BL (2008) The logopenic/phonological variant of primary progressive aphasia. Neurology 71:1227-1234 284. Spinelli EG, Mandelli ML, Miller ZA, Santos-Santos MA, Wilson SM, Agosta F, Grinberg LT, Huang EJ, Trojanowski JQ, Meyer M, Henry ML, Comi G, Rabinovici G, Rosen HJ, Filippi M, Miller BL, Seeley WW, Gorno-Tempini ML (2017) Typical and atypical pathology in primary progressive aphasia variants. Ann Neurol 81:430-443 285. Weintraub S, Teylan M, Rader B, Chan KCG, Bollenbeck M, Kukull WA, Coventry C, Rogalski E, Bigio E, Mesulam MM (2020) APOE is a correlate of phenotypic heterogeneity in Alzheimer disease in a national cohort. Neurology 94:e607-e612 286. Crutch SJ, Schott JM, Rabinovici GD, Murray M, Snowden JS, van der Flier WM, Dickerson BC, Vandenberghe R, Ahmed S, Bak TH, Boeve BF, Butler C, Cappa SF, Ceccaldi M, de Souza LC, Dubois B, Felician O, Galasko D, Graff-Radford J, Graff-Radford NR, Hof PR, Krolak-Salmon P, Lehmann M, Magnin E, Mendez MF, Nestor PJ, Onyike CU, Pelak VS, Pijnenburg Y, Primativo S, Rossor MN, Ryan NS, Scheltens P, Shakespeare TJ, Suarez Gonzalez A, Tang-Wai DF, Yong KXX, Carrillo M, Fox NC (2017) Consensus classification of posterior cortical atrophy. Alzheimers Dement 13:870-884 287. Sahoo A, Bejanin A, Murray ME, Tosakulwong N, Weigand SD, Serie AM, Senjem ML, Machulda MM, Parisi JE, Boeve BF, Knopman DS, Petersen RC, Dickson DW, Whitwell JL, Josephs KA (2018) TDP-43 and Alzheimer's disease pathologic subtype in non-amnestic Alzheimer's disease dementia. J Alzheimers Dis 64:1227-1233 288. Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, van Swieten JC, Seelaar H, Dopper EG, Onyike CU, Hillis AE, Josephs KA, Boeve BF, Kertesz A, Seeley WW, Rankin KP, Johnson JK, Gorno-Tempini ML, Rosen H, Prioleau-Latham CE, Lee A, Kipps CM, Lillo P, Piguet O, Rohrer JD, Rossor MN, Warren JD, Fox NC, Galasko D, Salmon DP, Black SE, Mesulam M, Weintraub S, Dickerson BC, Diehl-Schmid J, Pasquier F, Deramecourt V, Lebert F, Pijnenburg Y, Chow TW, Manes F, Grafman J, Cappa SF, Freedman M, Grossman M, Miller BL (2011) Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134:2456-2477 289. Sakae N, Josephs KA, Litvan I, Murray ME, Duara R, Uitti RJ, Wszolek ZK, van Gerpen J, Graff-Radford NR, Dickson DW (2019) Clinicopathologic subtype of Alzheimer's disease presenting as corticobasal syndrome. Alzheimers Dement 15:1218-1228 290. Josephs KA, Whitwell JL, Tosakulwong N, Weigand SD, Murray ME, Liesinger AM, Petrucelli L, Senjem ML, Ivnik RJ, Parisi JE, Petersen RC, Dickson DW (2015) TAR DNA-binding protein 43 and pathological subtype of Alzheimer's disease impact clinical features. Ann Neurol 78:697-709 291. Ohm DT, Fought AJ, Martersteck A, Coventry C, Sridhar J, Gefen T, Weintraub S, Bigio E, Mesulam MM, Rogalski E, Geula C (2020) Accumulation of neurofibrillary tangles and activated microglia is associated with lower neuron densities in the aphasic variant of Alzheimer's disease. Brain Pathol 292. Jellinger KA, Attems J (2007) Neuropathological evaluation of mixed dementia. J Neurol Sci 257:80-87 293. Kapasi A, DeCarli C, Schneider JA (2017) Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol 134:171-186 294. Power MC, Mormino E, Soldan A, James BD, Yu L, Armstrong NM, Bangen KJ, Delano-Wood L, Lamar M, Lim YY, Nudelman K, Zahodne L, Gross AL, Mungas D, Widaman KF, Schneider J (2018) Combined neuropathological pathways account for age-related risk of dementia. Ann Neurol 84:10-22 295. Rahimi J, Kovacs GG (2014) Prevalence of mixed pathologies in the aging brain. Alzheimers Res Ther 6:82 296. Matej R, Tesar A, Rusina R (2019) Alzheimer's disease and other neurodegenerative dementias in comorbidity: A clinical and neuropathological overview. Clin Biochem 73:26-31 297. Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, Caswell C, Van Deerlin VM, Yan N, Yousef A, Hurtig HI, Siderowf A, Grossman M, McMillan CT, Miller B, Duda JE, Irwin DJ, Wolk D, Elman L, McCluskey L, Chen-Plotkin A, Weintraub D, Arnold SE, Brettschneider J, Lee VM, Trojanowski JQ (2018) Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 141:2181-2193 298. Thomas DX, Bajaj S, McRae-McKee K, Hadjichrysanthou C, Anderson RM, Collinge J (2020) Association of TDP-43 proteinopathy, cerebral amyloid angiopathy, and Lewy bodies with cognitive impairment in individuals with or without Alzheimer's disease neuropathology. Sci Rep 10:14579 299. Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA (2018) Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol 83:74-83 300. Schneider JA, Arvanitakis Z, Bang W, Bennett DA (2007) Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69:2197-2204 301. Jellinger KA (2006) Clinicopathological analysis of dementia disorders in the elderly--an update. J Alzheimers Dis 9:61-70 302. Wang BW, Lu E, Mackenzie IR, Assaly M, Jacova C, Lee PE, Beattie BL, Hsiung GY (2012) Multiple pathologies are common in Alzheimer patients in clinical trials. Can J Neurol Sci 39:592-599 303. McAleese KE, Walker L, Erskine D, Johnson M, Koss D, Thomas AJ, Attems J (2020) Concomitant LATE-NC in Alzheimer's disease is not associated with increased tau or amyloid-beta pathological burden. Neuropathol Appl Neurobiol online Sep 8: doi 10.1111/nan.12664 304. McAleese KE, Walker L, Erskine D, Attems J (2020) The impact of concomitant LATE-NC on hyperphosphorylated-s pathology and cognitive decline in Alzheimer's disease (abstract). Neuropathol Appl Neurobiol 46 (Suppl. 1):37 305. Jang H, Kim HJ, Sim Choe Y, Kim SJ, Park S, Kim Y, Woon Kim K, Hyoung Lyoo C, Cho H, Hoon Ryu Y, Choi JY, DeCarli C, Na DL, Won Seo S (2020) The impact of amyloid-beta or tau on cognitive change in the presence of severe cerebrovascular disease. J Alzheimers Dis 306. Malek-Ahmadi M, Perez SE, Chen K, Mufson EJ (2020) Braak stage, cerebral amyloid angiopathy, and cognitive decline in early Alzheimer's disease. J Alzheimers Dis 74:189-197 307. Robinson AC, Roncaroli F, Chew-Graham S, Davidson YS, Minshull J, Horan MA, Payton A, Pendleton N, Mann DMA (2020) The contribution of vascular pathology toward cognitive impairment in older individuals with intermediate Braak stage tau pathology. J Alzheimers Dis 77:1005-1015 308. Gauthreaux K, Bonnett TA, Besser LM, Brenowitz WD, Teylan M, Mock C, Chen YC, Chan KCG, Keene CD, Zhou XH, Kukull WA (2020) Concordance of clinical Alzheimer diagnosis and neuropathological features at autopsy. J Neuropathol Exp Neurol 79:465-473 309. Tolnay M, Sergeant N, Ghestem A, Chalbot S, De Vos RA, Jansen Steur EN, Probst A, Delacourte A (2002) Argyrophilic grain disease and Alzheimer's disease are distinguished by their different distribution of tau protein isoforms. Acta Neuropathol 104:425-434 310. Togo T, Cookson N, Dickson DW (2002) Argyrophilic grain disease: neuropathology, frequency in a dementia brain bank and lack of relationship with apolipoprotein E. Brain Pathol 12:45-52 311. Wurm R, Klotz S, Rahimi J, Katzenschlager R, Lindeck-Pozza E, Regelsberger G, Danics K, Kapas I, Bíró ZA, Stögmann E, Gelpi E, Kovacs GG (2020) Argyrophilic grain disease in individuals younger than 75 years: clinical variability in an underrecognized limbic tauopathy. Eur J Neurol May 13:doi: 10.1111/ene.14321 312. Ferrer I, Garcia MA, Gonzalez IL, Lucena DD, Villalonga AR, Tech MC, Llorens F, Garcia-Esparcia P, Martinez-Maldonado A, Mendez MF, Escribano BT, Bech-Serra JJ, Sabido E, de la Torre Gomez C, Del Rio JA (2018) Aging-related tau astrogliopathy (ARTAG): not only tau phosphorylation in astrocytes. Brain Pathol 28:965-985 313. Kovacs GG (2020) Astroglia and tau: new perspectives. Front Aging Neurosci 12:96 314. Wiersma VI, van Ziel AM, Vazquez-Sanchez S, Nölle A, Berenjeno-Correa E, Bonaterra-Pastra A, Clavaguera F, Tolnay M, Musters RJP, van Weering JRT, Verhage M, Hoozemans JJM, Scheper W (2019) Granulovacuolar degeneration bodies are neuron-selective lysosomal structures induced by intracellular tau pathology. Acta Neuropathol 138:943-970 315. Wiersma VI, Hoozemans JJM, Scheper W (2020) Untangling the origin and function of granulovacuolar degeneration bodies in neurodegenerative proteinopathies. Acta Neuropathol Commun 8:153 316. Hou X, Fiesel FC, Truban D, Castanedes Casey M, Lin WL, Soto AI, Tacik P, Rousseau LG, Diehl NN, Heckman MG, Lorenzo-Betancor O, Ferrer I, Arbelo JM, Steele JC, Farrer MJ, Cornejo-Olivas M, Torres L, Mata IF, Graff-Radford NR, Wszolek ZK, Ross OA, Murray ME, Dickson DW, Springer W (2018) Age- and disease-dependent increase of the mitophagy marker phospho-ubiquitin in normal aging and Lewy body disease. Autophagy 14:1404-1418 317. Koper MJ, Van Schoor E, Ospitalieri S, Vandenberghe R, Vandenbulcke M, von Arnim CAF, Tousseyn T, Balusu S, De Strooper B, Thal DR (2020) Necrosome complex detected in granulovacuolar degeneration is associated with neuronal loss in Alzheimer's disease. Acta Neuropathol 139:463-484 318. White LR, Edland SD, Hemmy LS, Montine KS, Zarow C, Sonnen JA, Uyehara-Lock JH, Gelber RP, Ross GW, Petrovitch H, Masaki KH, Lim KO, Launer LJ, Montine TJ (2016) Neuropathologic comorbidity and cognitive impairment in the Nun and Honolulu-Asia Aging Studies. Neurology 86:1000-1008 319. Jack CR, Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R (2018) NIA-AA Research Framework: Toward a biological definition of Alzheimer's disease. Alzheimers Dement 14:535-562 320. Nelson PT, Trojanowski JQ, Abner EL, Al-Janabi OM, Jicha GA, Schmitt FA, Smith CD, Fardo DW, Wang WX, Kryscio RJ, Neltner JH, Kukull WA, Cykowski MD, Van Eldik LJ, Ighodaro ET (2016) "New Old Pathologies": AD, PART, and cerebral age-related TDP-43 with sclerosis (CARTS). J Neuropathol Exp Neurol 75:482-498 321. Ricci M, Cimini A, Chiaravalloti A, Filippi L, Schillaci O (2020) Positron emission tomography (PET) and neuroimaging in the personalized approach to neurodegenerative causes of dementia. Int J Mol Sci 21:7481 322. Koychev I, Hofer M, Friedman N (2020) Correlation of Alzheimer disease neuropathologic staging with amyloid and tau scintigraphic imaging biomarkers. J Nucl Med 61:1413-1418
Copyright: © 2020 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |