|
|
|
Free Neuropathology 1:27 (2020) |
|
Original Paper |
|
Loss of Ramified Microglia Precedes Axonal Spheroid Formation in Adult-Onset Leukoencephalopathy with Axonal Spheroids. |
|
Murad Alturkustani 1,2,3, Qi Zhang 2,3, Basma AlYamany 2,3, Lee-Cyn Ang 2,3 |
|
1 Department of Pathology, King Abdulaziz University, Jeddah, Saudi Arabia |
|
Corresponding author: |
|
Submitted: 22 August 2020 Accepted: 9 September 2020 Copyedited by: Deanna C. Fang and Henry Robbert Published: 5 October 2020 |
|
Additional resources and electronic supplementary material: supplementary material |
|
Keywords: Adult-onset leukoencephalopathy with axonal spheroids, Leukodystrophy, Microglia, Axonal spheroids, White matter, HDLS, ALSP |
|
Abstract Two different pathological mechanisms have been suggested to underlie adult-onset leukoencephalopathy with axonal spheroids (ALAS). Pathological studies have suggested that ALAS involves primary axonopathy with secondary demyelination. However, the identification of mutations in Colony Stimulating Factor 1 Receptor (CSF1R), important for microglial survival, has suggested that ALAS is a microgliopathy. This study examines the correlation between microglial changes and axonopathy in ALAS. A total of 6 ALAS cases were studied. White matter lesions were classified into three evolving stages: 1) numerous axonal spheroids among well-myelinated fibers; 2) moderate loss of myelinated fibers with or without axonal spheroids; and 3) a leukodystrophy-like pattern of severe confluent axonal and myelin loss. Axonal spheroids and ramified microglia were semi-quantified and the lesions were assigned a score of 0–3. We found a strong correlation between the preponderance of axonal spheroids and ramified microglial loss. All areas with a predominance of axonal spheroids showed a near-complete absence of ramified microglia, which was also apparent in small cortical and white matter lesions. In contrast, some areas with no ramified microglia showed no axonal pathology. Our findings support the suggestion that ramified microglia loss precedes axonal spheroids formation. This observation will help to better understand the pathogenesis of ALAS and suggests a protective role of microglia. Introduction Two different pathological mechanisms have been suggested to underlie adult-onset leukoencephalopathy with axonal spheroids (ALAS). Pathological studies have proposed a primary axonopathy with secondary demyelination (1, 2), while genetic/molecular studies have identified a mutation in a gene important for microglial function and, hence, implied that ALAS is a microgliopathy (3). Moreover, a recent pathological study has identified a microglial morphological alteration (i.e. “dysplastic” microglia) as the pathological correlate for microgliopathy (4). ALAS is also known by other names in the literature with most common ones being hereditary diffuse leukoencephalopathy with spheroids (HDLS) and adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP). Recent advances in our understanding of the biology and functions of microglia have demonstrated both its beneficial and harmful effects on various diseases (5). Investigating the relationship between microgliopathy and axonopathy in the pathogenesis of ALAS may provide better insight into the role of microglia in this disease. Recent attempts to explain the pathogenesis yielded different conclusions: 1) generalized colony stimulating factor 1 receptor (CSF1R) signaling impairment in CD68-immunopositive microglia is followed by multifocal axonal degeneration (6); 2) increase in ionizing calcium-binding molecule 1 (IBA1) and CD68-immunopositive cell numbers precedes axonal pathology (7); 3) uneven distribution of IBA1-immunopositive cells in the white matter precludes any firm conclusions regarding the role of these cells in the pathology (4); and 4) CSF1R mutations lead to aberrant microglia density and distribution, and regional loss of microglia (8). In this study, we investigated the relationship between axonal pathology and microglial changes in ALAS using the pathological staging system of ALAS proposed by Alturkustani et al. (1). This system focuses on the specific stages of the lesions rather than the overall pathological stage of the entire case at a point in time (7) and should provide a more precise correlation. Materials and Methods A formal autopsy consent was provided in all cases and the Western University research ethics board approved the study. A total of 6 ALAS cases were studied. Four cases were described before (cases 1–4) (1). Two additional cases of ALAS were referred to the London Health Sciences Center laboratory in 2013–2017. The brains were fixed and prepared as described before (1). Representative samples were submitted for microscopic examination and staining, and immunostaining procedures were performed as described previously (1). These procedures include Luxol fast blue (LFB) with hematoxylin and eosin (HE) staining. For immunohistochemistry, the primary antibodies used were anti-amyloid precursor protein (APP; Chemicon, Temecula, CA), anti-phosphorylated neurofilament (SMI31; Covance, Berkeley, CA), and anti-glial fibrillary acidic protein (Dako, Carpinteria, CA). Mouse monoclonal anti-HLA-DR antibody (1:200; clone CR3/43; Dako, Carpinteria, CA) was used to label microglial cells. Anti-ionized calcium binding adaptor molecule 1 (IBA1) antibody (1:500, 019-19741, WAKO) was stained in Dr. V. Wee Yong’s laboratory (Calgary, Canada) following their previously published protocol (9). The white matter lesions were classified into three evolving stages, as discussed in our previous work (1): 1) numerous axonal spheroids within well-myelinated fibers; 2) moderate loss of myelinated fibers with or without axonal spheroids; and 3) a leukodystrophy-like pattern with severe confluent axonal and myelin loss. Digital images of LFB-HE, APP, HLA-DR, IBA1, and SMI31 staining slides were obtained using the Aperio system. Particular areas of interest were demarcated on the computer screen of these digital images. At least five areas for each pathological stage from each case were examined. The pathological features, regardless of the size of the lesion, defined the pathological stages. Any lesion can range from very small areas (usually in stage 1 pathology) to very large (common in stage 3 pathology). A single area in the white matter could contain more than one pathological stage. Axonal spheroids and the HLA-DR-expressing cells were semi-quantified (see supplemental figure 1) and lesions were assigned a score of 0–3. The scoring for axonal spheroids was as follows: 0 = absence of axonal spheroids; 1 = few axonal spheroids (1–2/high power field; HPF); 2 = moderate number of axonal spheroids (3–5/HPF); and 3 = frequent axonal spheroids (6 or more/HPF). For the HLA-DR and/or IBA1-expressing cells, we subdivided them into ramified-phenotype and amoeboid-phenotype. Their numbers were scored in the following manner: 0 = no HLA-DR staining; 1= few cells (1–9/HPF); 2 = moderate number of cells (10–19/HPF); and 3 = frequent cells (20 or more/HPF). Several morphological shapes of microglia have been identified (10). However, for the sake of simplicity, in this study we divided microglia into two different types: 1) ramified microglia with a small cell body with branching processes radiating from the cell bodies, including a broad spectrum of reactive microglial shapes with varying lengths and thicknesses of processes and different cell body dimensions; and 2) amoeboid microglia with a macrophage-like or amoeboid phenotype characterized by a round cytoplasm with no processes. The white matter from the same autopsy tissue in the areas adjacent to the affected white matter and separate from it was used as control tissue to confirm the reliability of the immunostains (i.e. internal positive control) used in this study. As the autopsy brain tissue from relatively normal cases was not expected to show axonal spheroids, we did not use such controls in our study.
Results The clinical information for the first four cases has been discussed before (1) and is summarized in table 1. In general, clinical presentations were dependent on the location of the white matter areas affected, which were preferentially frontotemporal. Case 5 was a 57-year-old female who had a rapid 2-year progression of neurobehavioral symptoms (self-neglect and apathy) and seizures until death. She had a sister with cognitive impairments, but slower progression. Both patients had a previously unreported variant of CSF1R (c.2563 C>T) mutation (11). Their mother had a history of early-onset dementia. Case 6 was a 38-year-old female with a rapid decline in cognition and short- and long-term memory impairment. She was placed in a nursing home as she required assistance for daily life activities and died two years after the initial presentation. Her first-degree relatives had a positive history of dementia. She had another novel variant of CSF1R (c.2377A>G) mutation. The CSF1R gene mutational analysis for the first 4 cases was unsuccessful as the quality of the DNA extracted from fixed tissue was inadequate, as discussed before (1). Both mutations were analyzed via the VarSome website (https://varsome.com/variant/hg19/NM_005211.3%3Ac.2563C%3ET and https://varsome.com/variant/hg19/NM_005211.3%3Ac.2377A%3EG). Various software tools including DANN (score 0.9991 and 0.9989, respectively), MutationTaster (disease causing), FATHMM (Damaging), FATHMM-MKL (Damaging), MetaSVM (Damaging), and MetaLR (Damaging) predicted pathogenicity, which was supported by locations within the protein kinase functional domain. Antibodies against HLA-DR and IBA1 are considered the most sensitive markers for microglia as both stain all the morphological forms of activated microglia, phagocytic, and dystrophic microglia (5). Although IBA1 may be considered to be a more sensitive marker for these microglia by some authors (5), we found that HLA-DR is more sensitive for microglia staining in the white matter and comparable to IBA1 staining in the gray matter. IBA1 immunostaining also showed background staining, requiring careful examination to distinguish the immunopositive cells from the background (see supplemental figure 2). This resulted in an inferior quality of images for analysis. The following results were confirmed by both HLA-DR and IBA1 immunostains but are better demonstrated with the HLA-DR images, which we will utilize in the rest of this article. HLA-DR and IBA1 immunostaining showed a wide variation of microglial morphology in both gray and white matter in all cases. Microglial processes showed variations in number (single to multiple), thickness, and length. Some of the processes showed discontinuous staining and some areas showed only fragmented processes, which could be considered dysplastic (4) or dystrophic (5). Microglia with fragmented processes were observed along with those with intact processes in areas of normal-appearing white matter. This wide morphological variation made it difficult to correlate between microglial morphology and axonal spheroids. Therefore, as indicated in Materials and Methods, we only considered microglia with the amoeboid phenotype (i.e. globular cytoplasm with no processes) as a separate form of microglia. All other morphological variations of microglia, including those with fragmented processes, were considered ramified/reactive microglia. Using this simplified categorization of HLA-DR and IBA1-expressing cells (ramified vs. amoeboid phenotype), we consistently detected the following changes in all 6 cases (table 2): the white matter lesions classified as stage 1 pathology showed well-myelinated areas with moderate to frequent (score 2–3) axonal spheroids and a consistent loss of HLA-DR-expressing cells with ramified morphology (score 0) in close proximity to the axonal spheroids (Figure 1A–F). Some areas had a few (score 1) HLA-DR-expressing microglia with amoeboid phenotype, while others showed none (score 0). This may indicate that these areas had a slightly longer pathology duration than the areas without these cells. The white matter adjacent to the affected areas was unremarkable, with no axonal spheroids, no pigmented microglia, or other abnormality (Figure 1G). However, HLA-DR immunostaining showed denser forms of “ramified” microglia (Figure 1H). The remarkable association of axonal spheroids and the complete loss of ramified microglia was also evident in the affected cortex (Figure 2A–D). This association was apparent even in a very small area of cortical involvement, while the immediately adjacent area showed ramified microglia (Figure 2E–H).
In stage 2 pathology, white matter showed reduced myelin staining (Figure 3A–B), a variable number of axonal spheroids ranging from none to frequent (score 0–3), moderate to frequent amoeboid microglia (score 2–3), and none to few ramified microglia. According to the distribution of axonal spheroids, these areas were subclassified into three components: 1) Areas with axonal spheroids (score 1–3), showing no ramified microglia (score 0), but moderate to frequent amoeboid microglia (score 2–3). These areas may represent an “active/ongoing injury.” The white matter surrounding these areas showed widespread dense ramified microglia (score 3), while cells with amoeboid-phenotype were present in the central area (score 2–3), and the surrounding white matter (Figure 3C–F), used as an internal control for HLA-DR immunostaining. 2) Areas with no axonal spheroids (score 0), few-moderate amoeboid microglia (score 1–2), and no ramified microglia (score 0; Figure 3G–H). These can be considered “susceptible areas” and can have a different degree of myelinated axons loss. 3) Areas with no axonal spheroids (score 0), few-moderate amoeboid microglia (score 1–2), and none to few ramified microglial cells (score 1). These may represent areas of secondary axonal degeneration.
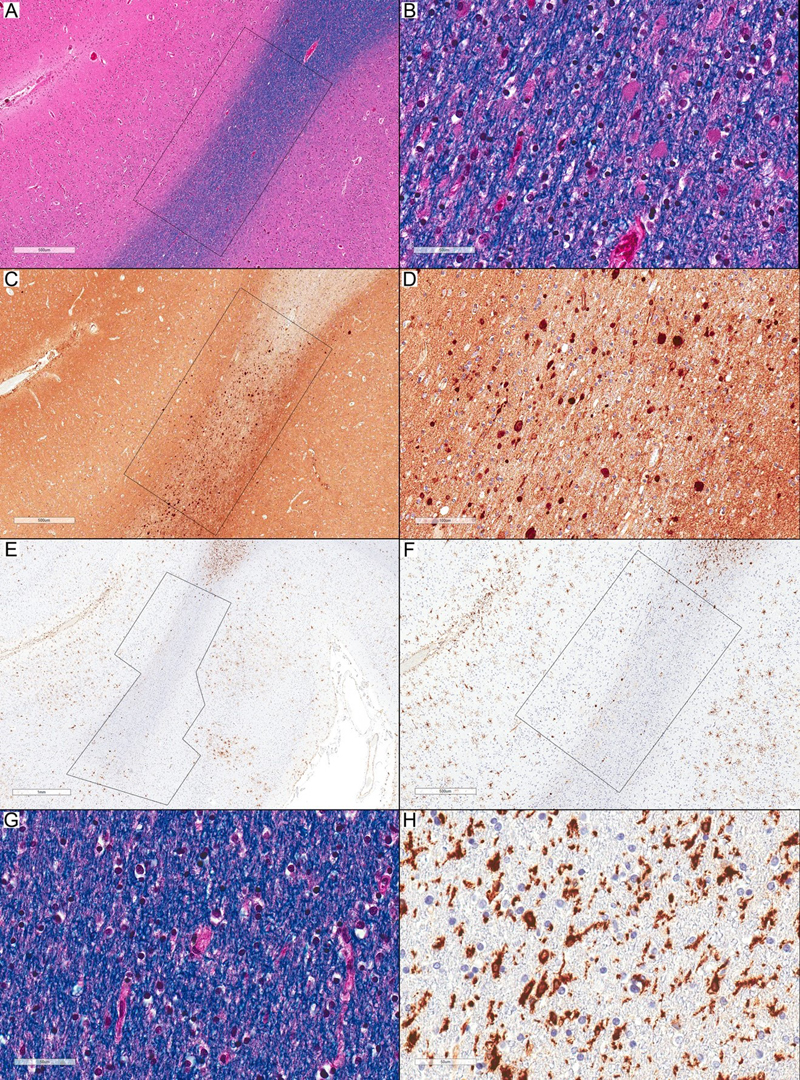
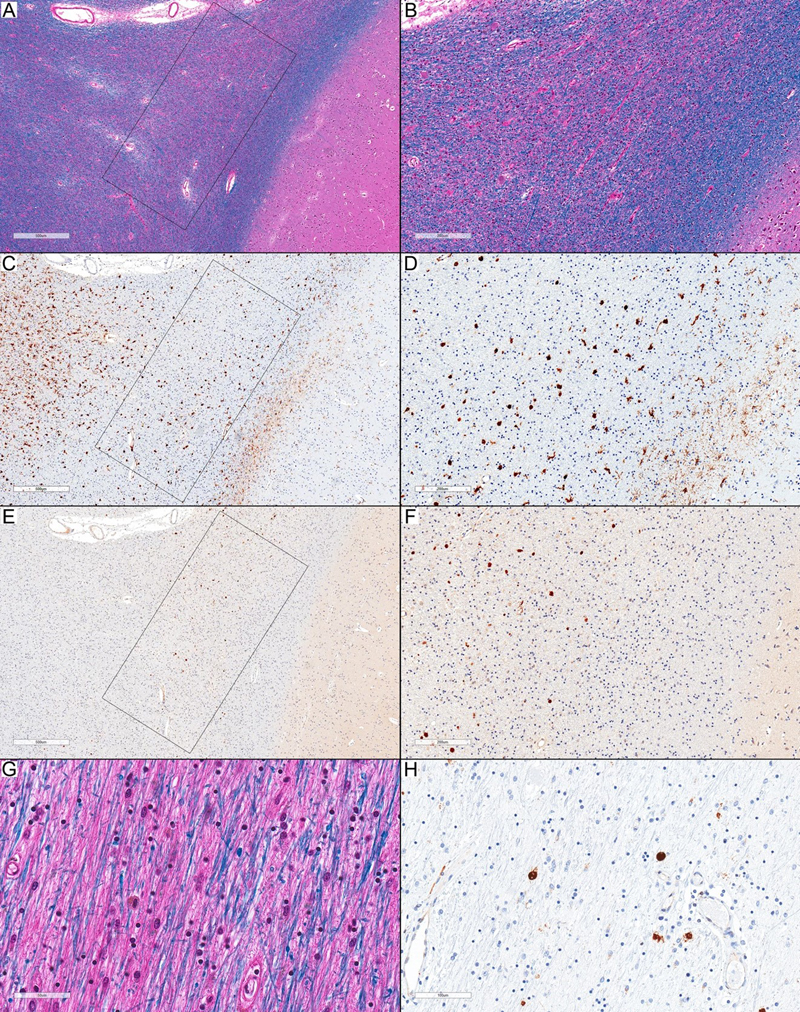
Figure 1. Microglia and axonal spheroids in Stage 1 pathology. (A) Normal-appearing white matter with frequent spheroids inside the rectangular inset (LFB-HE; scale bar: 500 µm). (B) Higher magnification of (A) shows many spheroids and no pigmented microglia (LFB-HE; scale bar: 50 µm). (C) APP immunostaining highlights frequent axonal spheroids inside the rectangular inset (APP; scale bar: 500 µm). (D) Higher magnification of (C) (APP; scale bar: 100 µm). (E) The affected area in (C) with many axonal spheroids in the white matter and a small area of adjacent cortex that shows a nearly complete loss of HLA-DR-expressing ramified microglia (inside the polygonal inset) and only a few scattered HLA-DR-expressing amoeboid-phenotype cells. The adjacent normal-appearing areas show an increased expression of HLA-DR-positive ramified microglia (HLA-DR; scale bar: 1 mm). (F) Higher magnification of (E) (HLA-DR; scale bar: 500 µm). (G) Normal-appearing white matter adjacent to the affected area shows no recognizable abnormalities (LFB-HE; scale bar: 50 µm). (H): HLA-DR immunostaining shows a frequent expression of HLA-DR-immunopositive ramified microglia (HLA-DR; scale bar: 50 µm). All images in this figure are representative of Case 2.
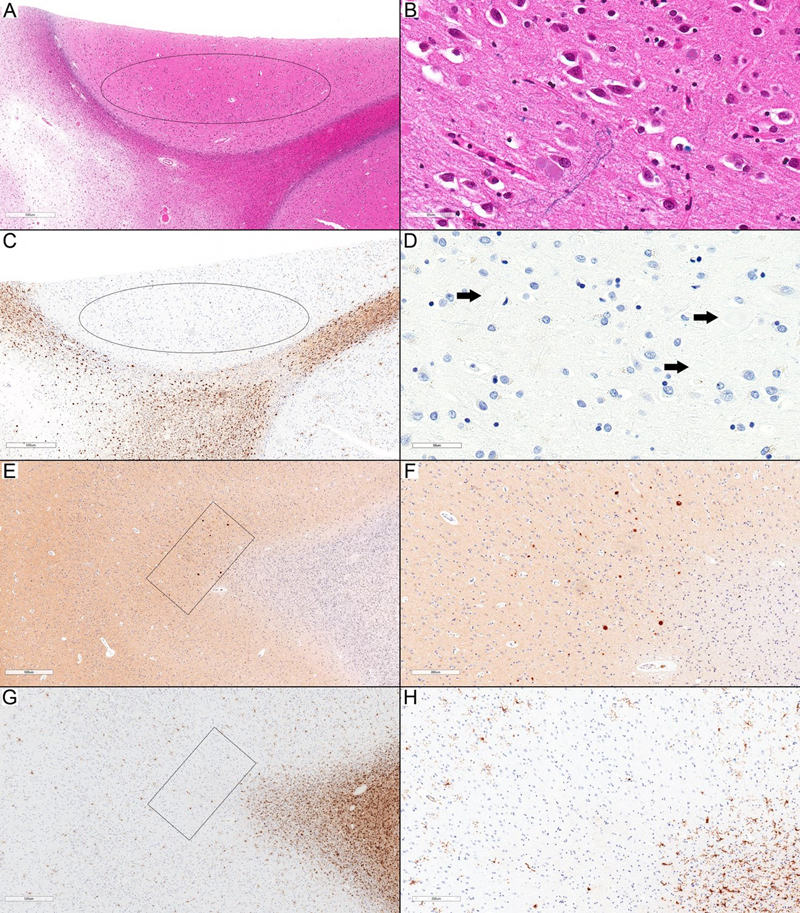
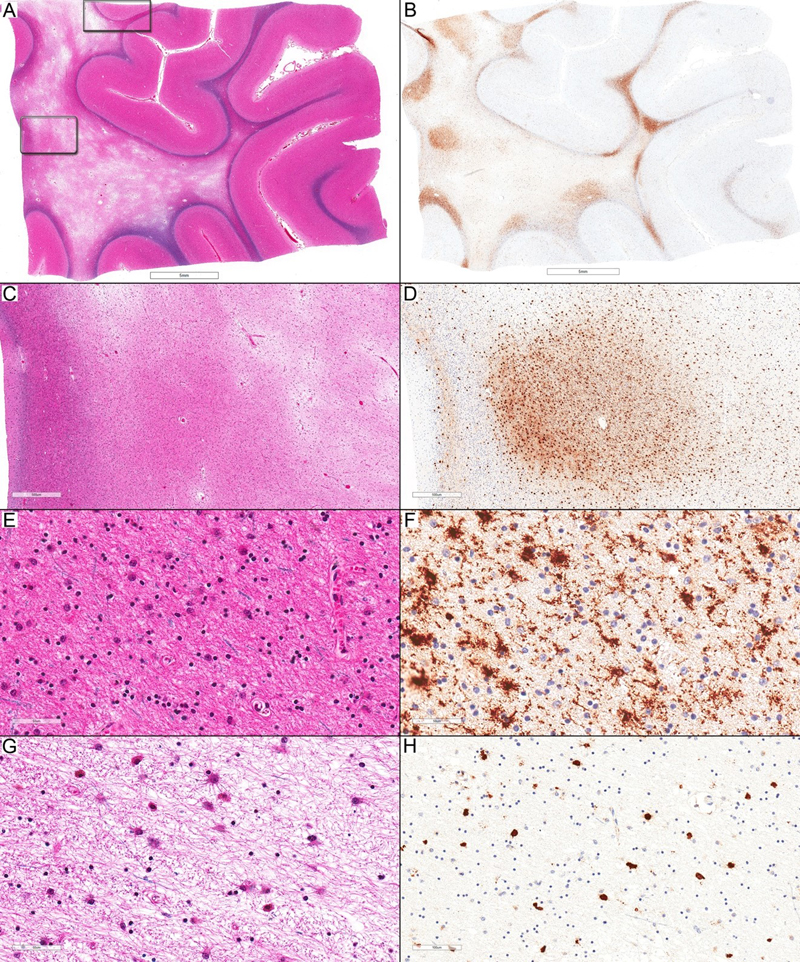
Figure 2. Cortical involvement in ALAS. (A) Focal involvement of the upper cortex with frequent axonal spheroids (inside ellipse) compared to the cortex on the right side. The underlying U-fibers are also affected (LFB-HE; scale bar: 500 µm). (B) Higher magnification of (A) from the cortical area inside the ellipse (LFB-HE; scale bar: 50 µm). (A) and (B) represent an inset (higher magnification of the upper rectangle) in Figure 4A. (C) HLA-DR immunostaining shows a complete focal loss of staining in the area with frequent axonal spheroids (inside ellipse) (HLA-DR; scale bar: 500 µm). (D) Higher magnification of (C) to highlight the presence of axonal spheroids (arrows) associated with a complete absence of HLA-DR-expressing ramified microglia (HLA-DR; scale bar: 50 µm). (E): A small cortical area (inside the rectangle) with frequent axonal spheroids (APP; scale bar: 500 µm). (F) Higher magnification of (E) (APP; scale bar: 50 µm.). (G) HLA-DR loss is limited to the very small affected area (inside the rectangle), while the surrounding cortical areas show HLA-DR-expressing ramified microglia (HLA-DR; scale bar: 500 µm). (H) Higher magnification of (G) (HLA-DR; scale bar: 200 µm). All images in this figure are representative of Case 6.
Figure 3. Microglia and axonal spheroids in Stage 2 pathology. (A) Decreased myelin staining in the deep subcortical white matter (mostly inside the inset) compared to the surrounding subcortical U-fibers (LFB-HE, scale bar: 500 µm). (B) Higher magnification of (A) (LFB-HE;, scale bar: 200 µm). (C) The area showing decreased myelin staining in (A) shows a complete loss of HLA-DR-expressing ramified microglia (mostly inside the inset) but moderate numbers of HLA-DR-immunopositive cells with amoeboid phenotype. The adjacent white matter shows frequent HLA-DR-expressing ramified microglia (HLA-DR; scale bar: 500 µm). (D) Higher magnification of (C) (scale bar: 200 µm). (E) APP-immunopositive axonal spheroids are limited to the area (mostly inside the inset) showing an extensive loss of ramified microglia and presence of HLA-DR expressing cells with amoeboid phenotype (APP; scale bar: 500 µm). (F) Higher magnification of (E) (APP; scale bar: 200 µm). (G): Another area with decreased myelin staining due to a prominent loss of myelinated fibers but without axonal spheroids (LFB-HE; scale bar: 50 µm). (H) Scattered HLA-DR-immunopositive cells with amoeboid phenotype in the same area (HLA-DR; scale bar: 100 µm). All images in this figure are representative of Case 5. In stage 3 pathology, the white matter showed a marked loss of myelinated axons and a relative preservation of the subcortical U-fibers (Figure 4A), which was classified as a “leukodystrophy-like” pattern. Although the white matter in this area as a whole was considered stage 3 pathology, HLA-DR immunostaining highlighted different patterns of staining (Figure 4B), which allowed us to appreciate variations in the preservation of the white matter and the distribution of HLA-DR-expressing cells in stage 3. We could observe areas of compact neuropil in the background, few small myelinated axons, a moderate number of oligodendrocytes, and mild to moderate gliosis (Figure 4C–E) along with rarefied areas showing a marked loss of oligodendrocytes and moderate gliosis (Figure 4G). HLA-DR immunostaining highlighted dense (score 3) ramified microglia in the areas showing higher white matter preservation and in the relatively preserved myelinated subcortical U-fibers (Figure 4B, D, F), while rarefied areas showed no ramified microglia (score 0) and a few to moderate (score 1–2) amoeboid microglia (Figure 4H). Axonal spheroids were absent (score 0) in most areas, yet scattered and rare (score 1) in others. Unfortunately, the imaging characteristics are not available for the radiological-pathological correlation. In general, stage 3 pathology, which denotes long-standing lesions, are prominent in frontal and temporal lobes, less prominent in parietal lobes, and rare in occipital lobes. Discussion Axonal spheroids and ramified microglia We found a strong association between the presence of axonal spheroids and a complete loss of ramified microglia in all pathological stages of ALAS lesions. This association was also apparent in very small areas with frequent axonal spheroids and in focal cortical lesions, strengthening the reliability of the findings. Although this was a semi-quantitative study, our findings demonstrate a strong correlation between the complete absence of ramified microglia (score 0) and the presence of axonal spheroids in affected areas. This finding could be considered “quantitative”, as score 0 denotes a complete absence of the ramified microglia. In stage 3, the presence of ramified microglia was associated with areas of better white matter preservation. These findings provide a link between pathological features observed (i.e. axonopathy) and the genetic abnormality identified (i.e. microgliopathy) in ALAS, supporting the beneficial effects of the reactive ramified microglia in maintaining axonal integrity. CSF1R and ALAS Identifying CSF1R mutations as the underlying genetic abnormality in ALAS suggested microgliopathy as the underlying abnormality (3, 12) in ALAS. As it is the most common mutation in ALAS, Konno et al. proposed the term “CSF1R-related leukoencephalopathy” for this entity (13). As the pathological features are the gold standard for ALAS diagnosis and not all ALAS cases have CSF1R alterations, this term would be specific to cases with confirmed CSF1R mutations only and would not be suitable for all ALAS cases. AARS2 mutations have been described in cases suspected to represent ALAS, including one case with biopsy findings supportive of this diagnosis (14). However, we concur with Konno et al. that “further studies, including detailed analyses of autopsied brains, are required to characterize AARS2 associated leukoencephalopathy” (13). CSF1R is a tyrosine kinase receptor whose signaling is fundamental for microglial survival (15). Two different mechanisms have been proposed for CSF1R mutations to affect the function of microglia in patients with ALAS: 1) Haploinsufficiency mechanism that results in a loss of function of CSF1R (8, 16, 17), and 2) a dominant-negative model (18). It is still unclear what the lost functions are or what is constitutive of the dominant-negative effect on the microglia. Oosterhof et al. concluded that the haploinsufficiency resulted in the maldistribution of microglia that leads to their local loss (8). Their recent work showed that homozygous mutations in CSF1R could result in the complete absence of microglia in pediatric-onset leukodystrophy (19). Our results confirmed the regional loss of ramified microglia in ALAS and found that the axonal pathology is mostly limited to these areas, supporting the importance of microglia in ALAS’ pathogenesis and suggesting a protective role of ramified microglia.
Figure 4. Microglia and axonal spheroids in Stage 3 pathology. (A) Marked loss of myelinated fibers in the deep white matter with relative preservation of the subcortical U-fibers (LFB-HE; scale bar: 5 mm). (B) Variable degree of HLA-DR expression in the subcortical deep white matter with multifocal areas containing HLA-DR-expressing ramified microglia. These areas are found mainly in the subcortical U-fibers and small foci of the adjacent white matter. The deeper white matter shows only scattered HLA-DR-expressing ramified microglia (HLA-DR; scale bar: 5 mm). (C) Higher magnification of the area within the lower rectangle outlined in (A). The deep white matter shows a variable loss of myelinated fibers. Areas of prominent loss show marked rarefaction, while areas with better preserved myelinated fibers in the center show a more compact parenchyma (LFB-HE; scale bar: 500 µm). (D) A better-preserved central area in (C) shows a marked expression of HLA-DR ramified microglia, while rarefied areas show only scattered HLA-DR-expressing ramified microglia and moderate to frequent HLA-DR-expressing cells with amoeboid phenotype (HLA-DR; scale bar: 500 µm). (E) Higher magnification of the better-preserved area in (C). Although there is a marked loss of myelinated fibers, a few scattered myelinated fibers remain, neuropil appears more compacted, and the loss of oligodendrocytes is mild to moderate (LFB-HE; scale bar: 50 µm). (F) Higher magnification of the central area in (D) (HLA-DR; scale bar: 50 µm). (G) The rarefied white matter areas show only occasional myelinated fibers, the neuropil is rarefied and mainly formed by astrocytic processes, and oligodendrocytes loss is moderate to severe (LFB-HE; scale bar: 50 µm). (H) HLA-DR immunostaining of the area in (G) shows mainly microglia with the amoeboid phenotype (HLA-DR; scale bar: 100 µm). All images in this figure are representative of Case 6. Morphological correlation of microglia and axonal degeneration Tada et al. described “dysplastic” microglia in brain samples from six patients with confirmed CSF1R mutations (4). These dysplastic microglia were interpreted as the morphological manifestation of the CSF1R mutation, but no association with axonal spheroids was established. In this study, we observed various reactive microglia morphologies, including some that can be considered “dystrophic”/“dysplastic”. However, the significance of these changes was hard to interpret and to correlate with the pathology. A different sequence of events in the pathogenesis of ALAS has been suggested. Riku et al. concluded that broad microglial impairment and low CSF1R expression on microglia preceded multifocal axonal degeneration (6). Meanwhile, Oyanagi et al. concluded that microglia proliferation preceded axonal swelling and loss. This conclusion suggests that microglial proliferation rather than loss leads to pathology (i.e. microglia is harmful and not beneficial; 7), which stands in contrast to our conclusions. In this study, we concluded that microglial loss preceded axonal degeneration based on the following observations: 1) There is a consistent complete loss of ramified microglia (by both HLA-DR & IBA1 immunostains) in all areas with axonal spheroids. As we did not observe any area with axonal spheroids to have reactive ramified microglia, it is unlikely that axonal spheroids precede microglia loss. 2) There is a presence of “susceptible areas” in stage 2 (i.e. areas with no ramified microglia and no axonal spheroids). Ramified microglia loss in these areas are significant as reactive microglia should be present in areas with white matter pathology. The presence of microglia with amoeboid phenotypes in these areas confirm the reliability of microglia immunostaining in these sections. 3) There is a presence of morphologically abnormal microglia in normal-appearing white matter. Tada et al., using ultrastructural examination, considered these dysplastic (4). Methodological differences between the studies might explain the difference in conclusions between this study and previous studies. In this study we described the correlation between the axonal spheroids and ramified microglia in each lesion based on its stage rather than the overall brain pathology used by Oyanagi et al. The latter system is useful for the correlation of clinical findings and the duration of pathology, while the former is more suitable to evaluate the mechanisms of a multifocal disease showing lesions with different pathological stages. Similar limitations may also apply to the conclusion drawn by Tada et al. on the microglial distribution in ALAS patients. They described spatial differences in microglia distribution with areas showing reduced numbers of microglia and others showing microgliosis in both the cortex and the white matter. They concluded that this differential distribution interferes with any meaningful quantitative statistical study of microglia depending on the area examined (4). This conclusion is valid if all lesions are studied together as one stage but not specifically for the stage of the individual lesion as analyzed in this study. We found that this spatial distribution did correlate with the axonal pathology in ALAS. The second significant difference between the previous studies and our study is that the other authors did not distinguish between ramified microglia and those with amoeboid phenotype. Many morphological variations of microglia have been identified (10). Although morphology is not a reliable predictor of function, microglia with amoeboid phenotype is an exception as it is consistently associated with phagocytosis (10). Therefore, we suggest that at least the microglia with amoeboid phenotype should be distinguished from other phenotypes. Microglia functions and pathology Microglia are regarded as immune cells of the central nervous system (CNS) and play a role in tackling viruses, phagocytosis, and as antigen-presenting cells (10, 20). Recently, additional functions have been demonstrated, such as active surveillance of synaptic integrity, regulation of brain development and potentially in blood vessel formation (10, 20). Microglia have been considered both pro-inflammatory and anti-inflammatory as well as neurotoxic and neuroprotective depending on their functional profiles (10, 20). The beneficial effects of microglia described include limiting the effects of cerebral ischemia in animal models since their absence through CSF1R blockade results in increased infarct size (21–23), while microglial repopulation reverses this effect (23). Microglial loss due to absence of CSF1R affects brain development and results in olfactory deficits in mice (24). Most harmful effects of microglia are associated with the concept of neuroinflammation, especially in multiple sclerosis (25), and after traumatic brain injury (26, 27). However, the term and the concept of neuroinflammation should be used with extreme caution, as suggested by Graeber and Streit (20). Microglia and axonal pathology There are only few studies that deal with the association between the axonal pathology and microglial reaction. In the experimental micropig model for mild traumatic brain injury, Lafrenaye et al. found that activated microglia contacted the proximal segments of injured axons (i.e. axonal spheroids) within 6 hours of the induced injury (28). Lafrenaye et al. suggested that microglia activation is beneficial and may enhance axonal regeneration through this contact (29). In a human study, microglia activation and microglia nodules were associated with axonal swelling in the perilesional white matter. These changes were demonstrated in the periplaque white matter lesions in multiple sclerosis, in white matter areas adjacent to infarcts, and in traumatic brain injury (30). In this study, the preferred hypothesis was that microglial reaction/nodules resulted in the axonal damage (30). However, no evidence of the timing of the events was provided to support this conclusion. Both studies (29, 30) showed the close association of reactive microglia to the axonal spheroids in affected white matter areas. The interpretation is quite different as the latter study suggested that neuroinflammation (defined as a microglial reaction) resulted in the axonal pathology (30), while the former study found that microglia reaction is secondary to axonal injury and suggested a neuroprotective function to these activated microglia. Our study described a different association between the axonal pathology and the ramified microglia, which, to our knowledge, has not been described before, and in which axonal spheroids were associated with loss of ramified microglia. The remarkable restriction of axonal spheroids to these areas, best appreciated in the small early lesions (i.e. stage 1 pathology), suggests that the loss of microglia is protective in preventing this pathology. Pathogenesis of ALAS The focus on the pathogenesis of ALAS has changed in the literature since the recognition of CSF1R mutations in ALAS. Previous pathological studies focused on the role of axonal pathology in the pathogenesis. The most relevant conclusions were: 1) The pathological features of the affected white matter suggest that the main pathological process is axonal degeneration with secondary demyelination (1); 2) the recurring or ongoing nature of the pathology is supported by the gradual progression of the white matter pathologic processes (31, 32); and 3) a possible role of hypoxic-ischemic injuries in the pathogenesis of ALAS (32, 33). Studies after the discovery of CSF1R mutations in ALAS focused on the effect of these mutations on microglia and the morphological changes of microglia in ALAS. The most relevant conclusions from these studies include: 1) CSF1R mutations can affect microglia through loss of function (haploinsufficiency model; 17) or dominant-negative effect (18); 2) demonstration of dystrophic microglia as the possible pathological correlate of these mutations (4); 3) change of microglia density precedes the white matter pathology, but with contradicting results. Oyanagi et al. proposed that microglia proliferation preceded axonal degeneration (7), while Oosterhof et al. found that microglia loss preceded it (8); and 4) CSF1R haploinsufficiency may lead to the maldistribution of microglia and result in the regional loss (8). There are many different ways to associate the previous observations and conclusions about the lesions in ALAS with meaningful pathogenesis. We propose the following pathogenesis: microglia loss precedes the axonal pathology and most likely represents an early stage of the pathology. However, it is still unclear what causes axonal injuries to develop in these restricted areas (and how), and whether this progression occurs with time only (e.g. aging process) or if an additional etiology (e.g. hypoperfusion) is required. Axonal injuries that result in axonal spheroids formation have many causes, including hypoxic-ischemic, metabolic, toxic, and traumatic causes. In previous studies of ALAS, evidence of oxidative stress caused by hypoxic-ischemic etiology was presented (32, 33) and this may represent possible additional etiological factors in the pathogenesis of ALAS. The effects of hypoxic-ischemic injury depend on the severity and the duration of the hypoxic-ischemic events. As there are no morphological features of moderate to severe hypoxic-ischemic injury in ALAS, and the axonal injuries are limited to areas of microglia loss, we concluded that this should be viewed as a mild form of injury. One possible cause of mild hypoxic-ischemic events that occur with aging is age-related cerebral hypoperfusion (34), which could potentially explain the delayed presentation and progression of lesions in ALAS. We emphasize that this is speculative, and not a conclusion of this study, but it might be an explanation of the observed correlation between the axonal pathology and the microglial loss. However, this proposed pathogenesis indicates that progression of ALAS is potentially preventable through: 1) replacement of the lost ramified microglia to prevent further damage through their protective function, and 2) improving or preventing the reduced cerebral blood flow associated with aging. The first suggestion supports the promising results from allogeneic haematopoietic stem cell transplantation (to repopulate the microglial niche) in patients with ALAS (35, 36). The second suggestion is currently unpractical, even in the animal model, but this may change in the future. Limitation of the study The major limitation of this study was the inability to test for CSF1R mutation from the paraffin sections in four of the six cases. However, the histopathological diagnosis of ALAS is reliable and considered the current gold standard as new mutations in different genes are being discovered in ALAS. The pathological features of all cases were similar and the characteristic three pathological stages were present in all cases. There was no difference in the morphological features between cases with confirmed CSF1R alterations (case 5 and 6), and cases with no genetic confirmation (cases 1–4). The association of microglia distribution to the axonal spheroids and the pathological stages were similar in all cases, as well (table 2). Our results and conclusions in this study (i.e. the correlation between microglia loss and axonal spheroids) are based on the pathological findings only, regardless of the underlying genetic alterations. As such, this limitation should not depreciate our results and conclusions. However, it restricted our ability to study the correlation between CSF1R mutation and the morphological changes. Conclusions The main finding of this study is a multifocal loss of ramified microglia in ALAS. This loss was seen in the affected areas of all pathological stages and best correlated with the presence of axonal spheroids. The presence of dense ramified microglia was associated with better preservation of the white matter. We concluded that ramified microglia most likely have a protective function for the white matter and that their absence contributes to axonal pathology observed in ALAS. These findings will contribute to a better understanding of the pathogenesis of ALAS and the protective roles of microglia, thus providing possible areas of intervention to prevent the progression of the disease. Acknowledgments We are grateful for the contributions of Professor V. Wee Yong, University of Calgary and Hotchkiss Brain Institute, Alberta, Canada (performing IBA1 immunohistochemistry), the neuropathology technologists at the London Health Sciences Centre, Ontario, Canada (technical support), Dr. Ashraf Dallol, King Abdulaziz University, Jeddah, Saudi Arabia (interpretation of genetic data) and the Department of Pathology, Western University, Ontario, Canada (research funding for Lee-Cyn Ang). Part of this work was presented as a poster at the XIXth International Congress of Neuropathology, Tokyo, September 23–27, 2018. References 1. Alturkustani M, Keith J, Hazrati LN, Rademakers R, Ang LC. Pathologic staging of white matter lesions in adult-onset leukoencephalopathy/leukodystrophy with axonal spheroids. J Neuropathol Exp Neurol 2015:74;233-40. 2. Alturkustani M, Sharma M, Hammond R, Ang LC. Adult-onset leukodystrophy: review of 3 clinicopathologic phenotypes and a proposed classification. J Neuropathol Exp Neurol 2013:72;1090-103. 3. Rademakers R, Baker M, Nicholson AM, Rutherford NJ, Finch N, Soto-Ortolaza A, Lash J, Wider C, Wojtas A, DeJesus-Hernandez M, Adamson J, Kouri N, Sundal C, Shuster EA, Aasly J, MacKenzie J, Roeber S, Kretzschmar HA, Boeve BF, Knopman DS, Petersen RC, Cairns NJ, Ghetti B, Spina S, Garbern J, Tselis AC, Uitti R, Das P, Van Gerpen JA, Meschia JF, Levy S, Broderick DF, Graff-Radford N, Ross OA, Miller BB, Swerdlow RH, Dickson DW, Wszolek ZK. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet 2012:44;200-5. 4. Tada M, Konno T, Tada M, Tezuka T, Miura T, Mezaki N, Okazaki K, Arakawa M, Itoh K, Yamamoto T, Yokoo H, Yoshikura N, Ishihara K, Horie M, Takebayashi H, Toyoshima Y, Naito M, Onodera O, Nishizawa M, Takahashi H, Ikeuchi T, Kakita A. Characteristic microglial features in patients with hereditary diffuse leukoencephalopathy with spheroids. Ann Neurol 2016:80;554-65. 5. Streit WJ, Xue QS, Tischer J, Bechmann I. Microglial pathology. Acta Neuropathol Commun 2014:2;142. 6. Riku Y, Ando T, Goto Y, Mano K, Iwasaki Y, Sobue G, Yoshida M. Early pathologic changes in hereditary diffuse leukoencephalopathy with spheroids. J Neuropathol Exp Neurol 2014:73;1183-90. 7. Oyanagi K, Kinoshita M, Suzuki-Kouyama E, Inoue T, Nakahara A, Tokiwai M, Arai N, Satoh JI, Aoki N, Jinnai K, Yazawa I, Arai K, Ishihara K, Kawamura M, Ishizawa K, Hasegawa K, Yagisita S, Amano N, Yoshida K, Terada S, Yoshida M, Akiyama H, Mitsuyama Y, Ikeda SI. Adult onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) and Nasu-Hakola disease: lesion staging and dynamic changes of axons and microglial subsets. Brain Pathol 2017:27;748-69. 8. Oosterhof N, Kuil LE, van der Linde HC, Burm SM, Berdowski W, van Ijcken WFJ, van Swieten JC, Hol EM, Verheijen MHG, van Ham TJ. Colony-Stimulating Factor 1 Receptor (CSF1R) Regulates Microglia Density and Distribution, but Not Microglia Differentiation In Vivo. Cell Rep 2018:24;1203-17 e6. 9. Sarkar S, Doring A, Zemp FJ, Silva C, Lun X, Wang X, Kelly J, Hader W, Hamilton M, Mercier P, Dunn JF, Kinniburgh D, van Rooijen N, Robbins S, Forsyth P, Cairncross G, Weiss S, Yong VW. Therapeutic activation of macrophages and microglia to suppress brain tumor-initiating cells. Nat Neurosci 2014:17;46-55. 10. Boche D, Perry VH, Nicoll JA. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol 2013:39;3-18. 11. Taylor R, Alyamany B, Pandey S, Kertesz A, Ang LC, Finger E. Two Distinct Clinical Phenotypes in a Family with ALSP Caused by a Novel CSF-1R Mutation.(P2. 176). AAN Enterprises, 2018. 12. van der Knaap MS, Bugiani M. Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol 2017:134;351-82. 13. Konno T, Kasanuki K, Ikeuchi T, Dickson DW, Wszolek ZK. CSF1R-related leukoencephalopathy: A major player in primary microgliopathies. Neurology 2018:91;1092-104. 14. Lynch DS, Zhang WJ, Lakshmanan R, Kinsella JA, Uzun GA, Karbay M, Tufekcioglu Z, Hanagasi H, Burke G, Foulds N, Hammans SR, Bhattacharjee A, Wilson H, Adams M, Walker M, Nicoll JA, Chataway J, Fox N, Davagnanam I, Phadke R, Houlden H. Analysis of Mutations in AARS2 in a Series of CSF1R-Negative Patients With Adult-Onset Leukoencephalopathy With Axonal Spheroids and Pigmented Glia. JAMA Neurol 2016:73;1433-9. 15. Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, Green KN. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014:82;380-97. 16. Pridans C, Sauter KA, Baer K, Kissel H, Hume DA. CSF1R mutations in hereditary diffuse leukoencephalopathy with spheroids are loss of function. Sci Rep 2013:3;3013. 17. Konno T, Tada M, Tada M, Koyama A, Nozaki H, Harigaya Y, Nishimiya J, Matsunaga A, Yoshikura N, Ishihara K, Arakawa M, Isami A, Okazaki K, Yokoo H, Itoh K, Yoneda M, Kawamura M, Inuzuka T, Takahashi H, Nishizawa M, Onodera O, Kakita A, Ikeuchi T. Haploinsufficiency of CSF-1R and clinicopathologic characterization in patients with HDLS. Neurology 2014:82;139-48. 18. Hume DA, Caruso M, Ferrari-Cestari M, Summers KM, Pridans C, Irvine KM. Phenotypic impacts of CSF1R deficiencies in humans and model organisms. J Leukoc Biol 2019. 19. Oosterhof N, Chang IJ, Karimiani EG, Kuil LE, Jensen DM, Daza R, Young E, Astle L, van der Linde HC, Shivaram GM, Demmers J, Latimer CS, Keene CD, Loter E, Maroofian R, van Ham TJ, Hevner RF, Bennett JT. Homozygous Mutations in CSF1R Cause a Pediatric-Onset Leukoencephalopathy and Can Result in Congenital Absence of Microglia. Am J Hum Genet 2019:104;936-47. 20. Graeber MB, Streit WJ. Microglia: biology and pathology. Acta Neuropathol 2010:119;89-105. 21. Berezovskaya O, Maysinger D, Fedoroff S. Colony stimulating factor-1 potentiates neuronal survival in cerebral cortex ischemic lesion. Acta Neuropathol 1996:92;479-86. 22. Fedoroff S, Berezovskaya O, Maysinger D. Role of colony stimulating factor-1 in brain damage caused by ischemia. Neurosci Biobehav Rev 1997:21;187-91. 23. Szalay G, Martinecz B, Lenart N, Kornyei Z, Orsolits B, Judak L, Csaszar E, Fekete R, West BL, Katona G, Rozsa B, Denes A. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun 2016:7;11499. 24. Erblich B, Zhu L, Etgen AM, Dobrenis K, Pollard JW. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One 2011:6;e26317. 25. Lassmann H. Mechanisms of white matter damage in multiple sclerosis. Glia 2014:62;1816-30. 26. Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol 2011:70;374-83. 27. Cherry JD, Tripodis Y, Alvarez VE, Huber B, Kiernan PT, Daneshvar DH, Mez J, Montenigro PH, Solomon TM, Alosco ML, Stern RA, McKee AC, Stein TD. Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol Commun 2016:4;112. 28. Lafrenaye AD, Todani M, Walker SA, Povlishock JT. Microglia processes associate with diffusely injured axons following mild traumatic brain injury in the micro pig. J Neuroinflammation 2015:12;186. 29. Lafrenaye AD. Physical interactions between activated microglia and injured axons: do all contacts lead to phagocytosis? Neural Regen Res 2016:11;538-40. 30. Singh S, Metz I, Amor S, van der Valk P, Stadelmann C, Bruck W. Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathol 2013:125;595-608. 31. Yamashita M, Yamamoto T. Neuroaxonal leukoencephalopathy with axonal spheroids. Eur Neurol 2002:48;20-5. 32. Freeman SH, Hyman BT, Sims KB, Hedley-Whyte ET, Vossough A, Frosch MP, Schmahmann JD. Adult onset leukodystrophy with neuroaxonal spheroids: clinical, neuroimaging and neuropathologic observations. Brain Pathol 2009:19;39-47. 33. Ali ZS, Van Der Voorn JP, Powers JM. A comparative morphologic analysis of adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia--a role for oxidative damage. J Neuropathol Exp Neurol 2007:66;660-72. 34. Tarumi T, Zhang R. Cerebral blood flow in normal aging adults: cardiovascular determinants, clinical implications, and aerobic fitness. J Neurochem 2018:144;595-608. 35. Eichler FS, Li J, Guo Y, Caruso PA, Bjonnes AC, Pan J, Booker JK, Lane JM, Tare A, Vlasac I, Hakonarson H, Gusella JF, Zhang J, Keating BJ, Saxena R. CSF1R mosaicism in a family with hereditary diffuse leukoencephalopathy with spheroids. Brain 2016:139;1666-72. 36. Gelfand JM, Greenfield AL, Barkovich M, Mendelsohn BA, Van Haren K, Hess CP, Mannis GN. Allogeneic HSCT for adult-onset leukoencephalopathy with spheroids and pigmented glia. Brain 2020:143;503-11.
Copyright: © 2020 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated |