|
|
|
Free Neuropathology 1:19 (2020) |
|
Original Paper |
|
Treatment of autoimmune encephalomyelitis with a histone deacetylase inhibitor Analyzing the role of immune-response genes |
|
Arathi K. Jayaraman a, Karen Avgush a, Rashad Kulam a, Advait Soni a, Areeb Khan a, Mourad Kerdjoudj a, Sundararajan Jayaraman a,b |
|
a Dept. of Microbiology & Immunology, the University of Illinois at Chicago, 909 S Wolcott Avenue, Chicago, IL 60612, USA |
|
Corresponding author: |
|
Submitted: 28 May 2020 Accepted: 08 July 2020 Copyedited by: Laura Peferoen Published: 14 July 2020 |
|
Additional resources and electronic supplementary material: supplementary material |
|
Keywords: Autoreactive T cells, Epigenetic regulation, Gene expression, Experimental autoimmune encephalomyelitis, Trichostatin A, Multiple sclerosis |
|
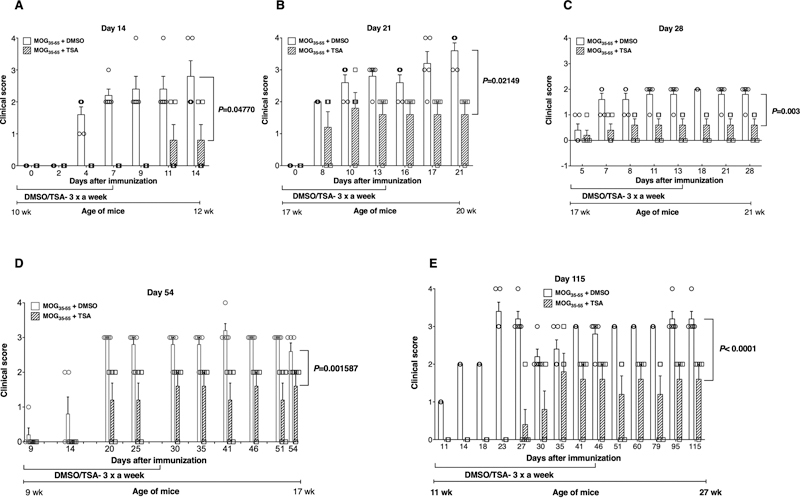
Abstract We have previously shown that treatment of female NOD mice with a potent nonselective histone deacetylase inhibitor attenuated experimental autoimmune encephalomyelitis, a model for progressive multiple sclerosis. Herein we show that immunization with the MOG35-55 peptide induced prolonged upregulation of genes encoding interleukin 17A (IL-17A), aryl hydrocarbon receptor, and histone deacetylase 11 in the spinal cord whereas the subunits of IL-27, IL-27p28 and IL-27ebi3 were significantly increased in secondary lymphoid organs after a lag period. Interestingly, the nitric oxide synthase gene was prominently expressed in both of these anatomic compartments following immunization. Treatment with the histone modifier repressed the transcription of all of these genes induced by immunization. Moreover, the drug suppressed the steady-state levels of the migration inhibitory factor and CD274 genes in both the spinal cord and peripheral lymphoid tissues. At the same time, the CD39 gene was downregulated only in secondary lymphoid organs. Paradoxically, the epigenetic drug enhanced the expression of Declin-1 in the spinal cord, suggesting a protective role in neuronal disease. Immunization profoundly enhanced transcription of the chemokine CCL2 in the secondary lymphoid tissues without a corresponding increase in the translation of CCL2 protein. Histone hyperacetylation neither altered the transcription of CCL2 nor its cognate receptor CCR2 in the central nervous system and peripheral lymphoid tissues. Surprisingly, the drug did not exert modulatory influence on most other immune response-related genes previously implicated in encephalomyelitis. Nevertheless, our data uncover several potential molecular targets for the intervention of experimental autoimmune encephalomyelitis that have implications for the treatment of progressive multiple sclerosis. 1. Introduction Multiple sclerosis (MS), an inflammatory disease of the central nervous system (CNS), manifests commonly as the relapsing-remitting disease. Some patients develop primary and secondary progressive forms of disease (Dendrou et al., 2015; Lassmann, 2017). The term ‘progressive multiple sclerosis’ has been proposed to encompass the primary and secondary progressive multiple sclerosis since there are more similarities than differences between them (Fox and Chataway, 2017). Although many drugs are effective in reducing relapses, they failed to reverse axonal degeneration and are sometimes associated with adverse side effects, including progressive multifocal leukoencephalopathy (Wingerchuk and Carter, 2014; Tintore et al., 2019). Thus, effective drugs for the treatment of progressive multiple sclerosis remain an unmet need. Since the early description of the experimental autoimmune encephalomyelitis (EAE) in monkeys (Rivers et al., 1933), monophasic, self-resolving, ‘classic’ EAE, as well as ‘atypical’ EAE, have been induced in rats and mice using whole spinal cord homogenates or peptides derived from the CNS components such as myelin oligodendrocyte glycoprotein (MOG), myelin basic protein (MBP) and proteolipid protein (PLP)(Dendrou et al., 2015; Lassmann and Bradl, 2017). Autoimmune-prone female non-obese diabetic (NOD) mice immunized with the MOG35-55 peptide consistently induced severe and long-lasting progressive EAE (PEAE) in 100% of animals characterized by paralysis of fore and hind limbs with (Slavin et al., 1998; Hidaka et al., 2014; Dang et al., 2015) or without discernible remissions (Basso et al., 2008; Jayaraman et al., 2017; 2018). Regardless, PEAE induced in NOD mice mimics features of progressive MS including the life-long disease, prominent demyelination, axonal loss, and astrogliosis (Slavin et al., 1998; Basso et al., 2008; Hidaka et al., 2014; Dang et al., 2015; Jayaraman et al., 2017; 2018), and hence is ideal for testing the efficacy of new drugs to treat progressive MS. The EAE model is amenable for the investigation of the genes critically involved in MS pathogenesis. Previous studies analyzed the roles of various immune response-related genes in EAE using gene knockout mice and specific neutralizing antibodies in wild-type mice. These studies investigated the role of T helper 1 (Th1), Th17, and Th1/Th17 (Th17.1) subsets as well as lymphokines such as interferon-γ (IFN- γ) (Ferber et al., 1996; Okuda et al., 1998; Hidaka et al., 2014), interleukin-4 (IL-4) (Falcone et al., 1998; Okuda et al., 1998; Ponomarev et al., 2007), IL-17A (Evangelidou et al., 2014), IL-10, colony-stimulating factor 1 (Borjini et al., 2016), IL-22 (Kreymborg et al., 2007), transforming growth factor-β (TGF-β (Okuda et al., 1998) and IL-27 (Li et al., 2005) in EAE. Similarly, the importance of the transcription factors T-bet (O’Connor et al., 2013), Gata-3 (Fernando et al., 2014), and RORγt (Martinez et al., 2014) respectively, involved in the transcription of IFN-γ, IL-4, and IL-17A, was also studied in EAE models. Moreover, the factors required for the induction of Th1 cells such as IL-12 (Gran et al., 2004), IL-18 (Lalor et al., 2011), and IL-23 (Kreymborg et al., 2007; El-Behi et al., 2011) critical for the generation of Th17 cells were implicated in EAE. The roles of the pluripotent cytokines, namely the tumor necrosis factor-α (TNF-α) (Hidaka et al., 2014; Borjini et al., 2016) and granulocyte-macrophage-colony-stimulating factor (GM-CSF/Csf2) (McQualter et al., 2001) were also studied in EAE. In addition to the adaptive immune system, the innate immune cells such as neutrophils (Jayaraman et al., 2018), and the CNS-resident microglia and astrocytes (Tran et al., 1997) appear to contribute to EAE. Interestingly, the first described and phylogenetically conserved pluripotent cytokine, the migration inhibitory factor (MIF) (Jayaraman and Muthukkaruppan, 1977), has been implicated in MS (Niino et al., 2000) and EAE (Powell et al., 2005). Other determinants critical for EAE include the orphan receptor, aryl hydrocarbon receptor (Ahr) (Nakahama et al., 2017), the transcription factors eomesodermin (Eomes) (Raveney et al., 2015) and Declin-1 (Dec1/Bhlhe40) (Lin et al., 2016), the immunoregulatory molecule CD39 (Mascanfroni et al., 2013), matrix metalloproteinases (MMP) (Kandagaddala et al., 2012; Rempe et al., 2016), and the chemokine CCL2 (Mahad et al., 2003; Moreno et al., 2014; Hidaka et al., 2014). The caveat is the lack of consensus regarding the critical gene(s) involved in EAE. Variations including the genetic background of experimental animals (rats and mice), types of EAE models studied (short-term, self-resolving ‘classic’ EAE, ‘atypical’ EAE, relapsing-remitting EAE, and PEAE), the immunogens used for EAE elicitation (whole spinal cord homogenates, peptides derived from MOG, MBP and PLP), tissues (CNS vs. peripheral lymphoid cells) and the time points (peak vs. chronic phase) examined can contribute to the uncertainty of the results. Hence, investigation of the roles of these various immune response-related genes in a well-characterized model is likely to yield crucial information on the impact of these genes on neurodegeneration. Our previous work demonstrated that the treatment of MOG35-55 immunized female NOD mice with the most potent histone deacetylase (HDAC) inhibitor Trichostatin A (TSA) (De Ruijter et al., 2003) improved the clinical symptoms of PEAE (Jayaraman et al., 2017). Protection from PEAE was accompanied by histone H3 hyperacetylation in the spinal cord (SC) and spleen, reduced influx of T cells, and neutrophils into the CNS as well as diminished axonal damage of the neurons in the CNS (Jayaraman et al., 2017; 2018). To gain insights into the roles of various genes in the PEAE model, we studied the expression profiles of 41 genes encoding lymphokines, transcription factors, accessory cell-associated determinants, and chemokines in the CNS and secondary lymphoid organs (SLO) longitudinally during the prolonged course of the disease (27-weeks). Surprisingly, only a small set of mostly non-overlapping genes were differentially upregulated in the CNS and SLO, which were substantially repressed by TSA treatment, indicating their possible roles in PEAE. These data suggest that similar perturbation of the epigenome of MS patients may facilitate the identification of molecular targets for the development of novel drugs to treat this debilitating disease. 2. Material and methods 2.1. EAE induction and treatment This study was approved by the Institutional Animal Care and Use Committee of The University of Illinois at Chicago and conducted according to the National Institutes of Health guide for the care and use of Laboratory Animals (NIH Publications No. 8023, revised 1978). Female NOD/ShiLtj mice were purchased from The Jackson Laboratories (Bar Harbor, ME) and immunized subcutaneously (s.c) on the flank with 100 µg of mouse MOG35-55 peptide (Tocris Bioscience) emulsified in complete Freund's adjuvant and pertussis toxin was administered intravenously (Jayaraman et al., 2017; 2018). Randomly chosen littermates were injected s.c on the flank with 500 µg of TSA (Sigma Chemical Company, St. Louis, MO) per Kg body weight three times a week. Controls received the same amount of the vehicle, dimethyl sulfoxide (DMSO) (Sigma) diluted in phosphate-buffered saline (PBS). The body weight, blood glucose levels, (Jayaraman et al., 2010; 2013; Jayaraman and Jayaraman, 2018; Patel et al., 2011) and clinical scores (Jayaraman et al., 2017; 2018) were recorded three times a week. The EAE score was assigned as follows: 0, healthy, 1, limp tail, 2, one hind limb weakness, 3, both hind limb weakness, 4, forelimb weakness, 5, paralysis, moribund or death (Jayaraman et al., 2017; 2018). Five mice per group were chosen based on our previous investigations (Jayaraman et al., 2017; 2018). The data are presented as the mean ± SEM for each time point of observation. 2.2. Gene expression analysis We analyzed the expression levels of 41 genes in the entire SC and SLO (spleen and the draining inguinal, popliteal, axillary and cervical lymph nodes) of mice that were immunized with MOG35-55 and treated with DMSO or TSA. We have investigated 60 mice (five mice treated with DMSO and five mice with TSA at six-time points) for the expression of genes in the SC and SLO concurrently. To analyze the effect of the drug on the basal level of gene transcription regardless of immunization, we treated separate groups of 10 unimmunized mice with TSA or DMSO and analyzed one day later since TSA acts within hours of treatment (Van Lint et al., 1996; De Ruijter et al., 2003). These data are indicated at the day 1-time point in all figures. On the other hand, treatment groups received DMSO or TSA starting from the day of immunization. Mice were perfused with PBS before the spinal cord was extracted to avoid peripheral blood contamination (Jayaraman et al., 2017; 2018). Total RNA was isolated from individual mice using TRIzol (Invitrogen, Carlsbad, CA). Since preliminary experiments indicated similar levels of expression of genes among different mice in each group, the RNA from five mice per group at each time point was pooled from identical tissues to minimize individual variability, as described previously (Jayaraman et al., 2013). The RNA was treated with Turbo DNase and converted to cDNA using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, Carlsbad, CA), as described earlier (Jayaraman et al., 2010; 2013; 2017; 2018; Patel et al., 2011). Real-time quantitative reverse-transcriptase mediated polymerase chain reaction (qRT-PCR) was performed using the Applied Biosystems ViiA7 Real-time PCR system. The cDNA equivalent to 100 ng of total RNA was used along with the 2X SYBR Green master mix in the qRT-PCR assay. The primer sets were designed, and specificity validated using the Primer3 and BLAST programs (ncbi.nim.nih.gov) and BiSearch Web Server tool (bisearch.enzim.hu). The Mif primer sets were purchased from the Integrated DNA Technologies (Coralville, IA), and the custom primers were synthesized at the same facility. Whereas the primer sets for Gapdh, Il4, Il17a, Il18, Ifng, Nos2, Tnfa, Tbet, Rorgt, and Gata3 were described previously (Jayaraman et al., 2010; 2013), other primer sets are listed in Supplementary Table 1. Every cDNA sample was analyzed in triplicate at each time point, and the expression level of any given gene was ascertained using Gapdh as the normalizer (Jayaraman et al., 2010; 2013; 2017; 2018; Patel et al., 2011) since it was not altered by TSA treatment in vitro (Van Lint et al., 1996). The same cDNA pool was analyzed for the expression levels of all 41 genes. The gene expression level was determined using the 2-∆∆CT method (Jayaraman et al., 2010; 2013; 2017; 2018; Patel et al., 2011). The data are represented as the mean ± SEM of triplicate technical repeats per time point. The outliers that deviated >10% from other data points were omitted, and the samples reanalyzed for gene expression. 2.3. ELISA Sera were collected from naïve and immunized mice with or without TSA treatment and assayed for CCL2 using the ELISA Ready-SET-Go kit (eBioscience, San Diego, CA). Samples were pooled from five mice per group, analyzed in duplicate, and expressed per mg of protein. 2.4. Statistics The statistical significance of clinical scores between control and TSA-treated groups was determined using the area under the ROC curve. The difference in gene expression was calculated using an unpaired two-tailed Student's t-test. ELISA data were analyzed for significance between groups by two-way ANOVA. The P-value of <0.05 was considered significant. GraphPad Prism 6.0 software (San Diego, CA) was used for all statistical analyses. 3. Results 3.1. TSA treatment reduced the clinical manifestation of PEAE We have reported earlier that after immunization with the MOG35-55 peptide the bodyweight declined transiently between 13 and 16 days, which steadily increased after that regardless of treatment with DMSO or TSA (Jayaraman et al., 2017). Since female NOD mice develop type 1 diabetes in an age-dependent manner, we routinely monitored blood glucose levels three times a week throughout the experiments. As reported earlier (Jayaraman et al., 2010; 2013; Jayaraman and Jayaraman, 2018; Patel et al., 2011), mice that were 9-11 weeks old (Fig. 1A, D-E) were normoglycemic at the time of immunization with MOG35-55 emulsified in complete Freund’s adjuvant and remained diabetes-free throughout the observation, 12-27 weeks of age (Fig. 1A, D-E). However, in two experiments, 40 and 60% of 17-week old mice immunized and treated with DMSO were found to be diabetic. TSA treatment reduced the diabetes incidence respectively to 0% and 20% when analyzed at 20 and 21 weeks of age (Fig. 1B-C). These results are consistent with our previous observations that the administration of complete Freund’s adjuvant alone could effectively prevent type 1 diabetes in younger female NOD mice. In contrast, similar treatment of older (>13-weeks) mice did not have the same potency in preventing diabetes (Jayaraman and Jayaraman, 2018). These results indicate that whereas the development of type 1 diabetes is age-dependent, induction of PEAE by immunization with the MOG35-55 peptide emulsified in complete Freund’s adjuvant is independent of the age and the glycemic status of mice.
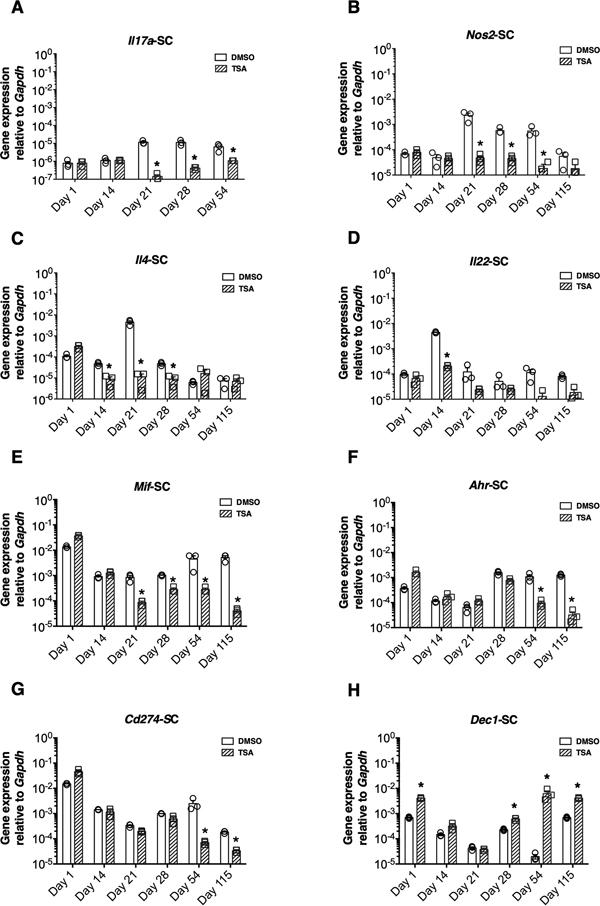
Fig. 1 TSA treatment improved clinical disease. All of the mice immunized with MOG35-55 developed PEAE without discernible remission (Fig. 1A-E), consistent with earlier reports (Basso et al., 2008; Jayaraman et al., 2017; 2018). The overall clinical severity was comparable in most instances except in a single experiment, probably due to the difference in experimental conditions (Fig. 1C). Treatment with TSA bestowed robust and irreversible protection from PEAE for the entire period of observation (115 days) even after the cessation of the drug administration on day 45 (Fig. 1E), as reported earlier (Jayaraman et al., 2017). Interestingly, the administration of the drug for one (Fig. 1A), two (Fig. 1B, C), four (Fig. 1D) or six-weeks (Fig. 1E) afforded a comparable level of protection against the disease. In previous studies, we have demonstrated that chronic TSA treatment decreased the influx of neutrophils and CD4+ T cells into the SC. Drug treatment also reduced the inflammation and particularly axonal damage in the spinal cord, indicating the neuroprotective effect of the HDAC inhibitor (Jayaraman et al., 2017; 2018). To understand the underlying mechanisms of drug-mediated protection against PEAE, in the current study, we have concurrently profiled the gene expression in the CNS and SLO at various time points shown in Fig. 1A-E. 3.2. Differential impact of the histone modifier on gene expression in the CNS The neuronal disease induced in NOD mice resembled the ‘classic’ EAE except that it lasted longer than in most strains of mice (Slavin et al., 1998; Basso et al., 2008; Hidaka et al., 2014; Dang et al., 2015; Jayaraman et al., 2017; 2018). Since the SC is thought to be the primary target of the ‘classic’ EAE and the brain damage was selectively observed in 'atypical’ EAE (Pierson and Goverman, 2017), we profiled the gene expression in the entire SC. Expression levels of genes encoding 11 lymphokines, four cytokines, seven accessory cell surface-associated determinants, seven transcription factors, and 11 histone deacetylases (Hdacs) were investigated by qRT-PCR. Unimmunized mice were separately treated with DMSO or TSA to determine the impact of drug treatment on the constitutive level of gene expression. The data are indicated at the day-1 time point in all figures. Mice immunized with MOG35-55 were treated with DMSO or TSA starting from the day of immunization. The entire data sets are presented in Supplementary Fig. 1-5. Only the differences in gene expression that occur consecutively at more than one-time point but not those altered transiently or sporadically are highlighted herein. Longitudinal analysis during chronic PEAE revealed that some immune response-related genes were induced upon MOG35-55 immunization after a 7-14-day lag period in both the CNS and SLO. The Th cell-associated Il17a, Il4, and Il22 and the inflammatory cytokine gene Nos2 were upregulated for a prolonged period between 21 and 54 days in the CNS in different experiments (Fig. 2A-D). The epigenetic drug suppressed the expression of these genes that were upregulated by immunization. Surprisingly, the constitutive expression of Mif, Ahr, and Cd274 was also notably repressed by TSA treatment during the chronic phase of the disease (Fig. 2E-G). Unexpectedly, the pan HDAC inhibitor upregulated the expression of Dec1 (Bhlhe40), specifically during the late stage of PEAE (Fig. 2H).
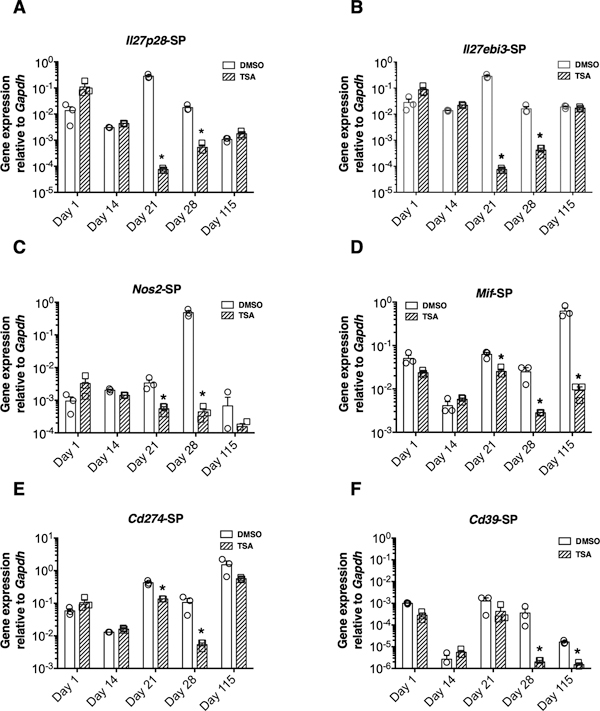
Fig. 2 Differential impact of the histone modifier on gene expression in the CNS. Although previous studies implicated many other genes in EAE, those encoding IL-23, IFN-γ, IL-18, IL-27p28, IL-27ebi3, IL-12p35, IL-10, GM-CSF, TNF-α, and TGF-β were not transcriptionally upregulated in the CNS of PEAE mice (see Supplementary Fig. 1-2). Similarly, the mRNA of the accessory cell surface-associated determinants, MMP9, MMP12, and CD74, remained stable in the CNS following immunization (Supplementary Fig. 3). Moreover, the transcription factor genes such as Tbet, Gata3, Rorgt, Eomes, and Foxp3 also remained unchanged in the SC (Supplementary Fig. 4). Notably, the histone modifier failed to alter the transcription of these genes. Thus, our comprehensive analysis uncovered the selective repression of genes in the CNS without affecting the levels of many other genes previously implicated in neuroinflammation. 3.3. The different patterns of gene regulation in the SLO by TSA treatment The expression of the genes encoding the subunits of the heterodimeric lymphokine IL-27 such as IL-27p28 and IL-27ebi3 peaked on day 21 and subsided steadily thereafter in the SLO (Fig. 3A-B). Treatment with TSA reduced the expression of these genes and the inflammatory gene Nos2 (Fig. 3C). Interestingly, the transcription of Mif, Cd274, and Cd39 in the SLO was also downregulated by the histone modifier (Fig. 3D-F). Surprisingly, the genes encoding the lymphokines such as IL-4, IL-10, IL-17A, IL-12p35, IL-18, IL-22, IL-23, IFN-γ, TGFβ, GM-CSF and TNF-α (Supplementary Fig. 1-2), as well as the macrophage-associated determinants, MMP9, MMP12, Arg-1, and CD74 were neither upregulated during pathogenesis nor repressed by TSA treatment in the SLO (Supplementary Fig. 3). Similarly, the transcription of Tbet, Rorgt, Gata3, Eomes, Dec1, Ahr, and Foxp3 remained mostly unaffected by the HDAC inhibitor treatment (Supplementary Fig. 4). Thus, the antigen-induced transcription of Il27p28, Il27ebi3, and Nos2, as well as the constitutive expression of Mif, Cd274, and Cd39, were selectively impeded by TSA treatment in the SLO. These data collectively demonstrate the differential influence of epigenetic modulation on gene expression in the CNS and SLO in the PEAE model.
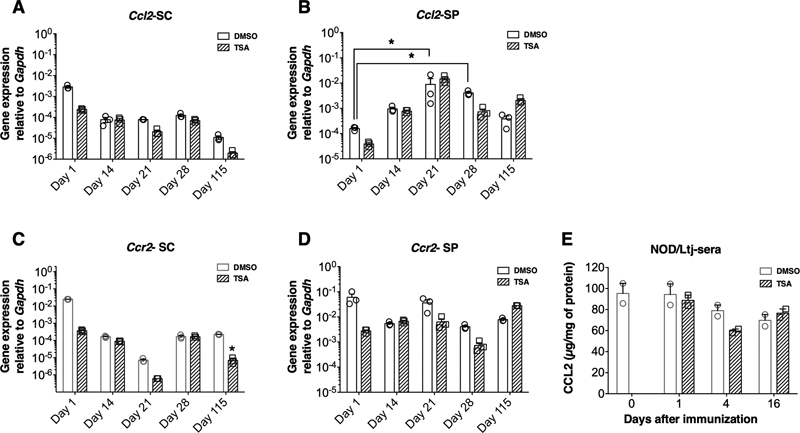
Fig. 3 Different patterns of gene regulation in the SLO by TSA treatment. 3.4. Epigenetic regulation failed to influence the prominent chemokine system Since previous work indicated the nonredundant roles of CCL2 and CCR2 in recruiting the inflammatory cells to the CNS (Mahad and Ransohoff, 2003; Moreno et al., 2014), we surmised that TSA-induced neuroprotection could also accompany modulation of these genes. Immunization of NOD mice with MOG35-55 did not transcriptionally upregulate Ccl2 in the CNS (Fig. 4A). On the other hand, Ccl2 transcription increased dramatically in the SLO between days 14 and 28 (Fig. 4B). However, TSA treatment failed to modulate the transcription of Ccl2 in the SC or SLO. The gene encoding the cognate receptor of CCL2, CCR2, was not distinctively upregulated in either the SC or SLO nor perturbed by the histone modifier (Fig. 4C-D). In contrast to the robust increase in the Ccl2 mRNA level in the SLO, the amount of CCL2 protein did not increase in circulation during the pre-symptomatic period (up to 16 days) as assessed by ELISA, which remained unaffected by TSA treatment (Fig. 4E). These data indicate that protection from PEAE afforded by the CNS-permeant TSA (Jayaraman et al., 2017; 2018) was not associated with the transcriptional regulation of the prominent chemokine system, CCL2: CCR2 either in the CNS or SLO.
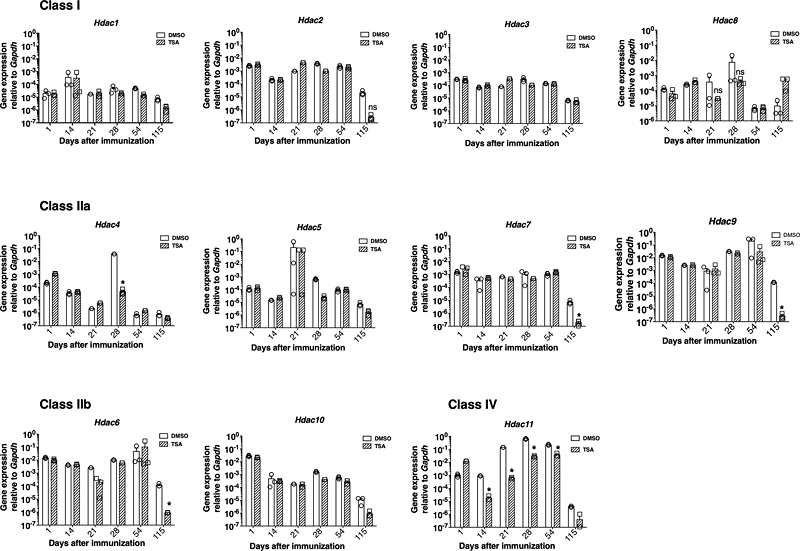
Fig. 4 Epigenetic regulation failed to influence the prominent chemokine system. 3.5. TSA treatment repressed the transcription of Hdac11 selectively in the CNS Whereas TSA can inhibit the activity of HDAC class I, IIa, and IIb isoenzymes with varying potency in vitro (Bradner et al., 2010), its ability to regulate Hdac genes in vivo has not been determined. To address this issue, we analyzed the mRNA levels of class I (Hdac1, Hdac2, Hdac3, and Hdac8), class IIa (Hdac4, Hdac5, Hdac7, and Hdac9), class IIb (Hdac6 and Hdac10), and class IV (Hdac11) HDACs using validated primer sets and qRT-PCR. Data shown in Fig. 5 indicate that immunization of NOD mice with MOG35-55 steadily increased the transcription of the class IV Hdac11 in the SC, which remained at high levels between 21 and 54 days postimmunization. Importantly, TSA treatment reduced the transcript level of Hdac11. Although Hdac1, Hdac4, Hdac5, Hdac6, Hdac8, and Hdac9 were modestly increased in the CNS with different kinetics after immunization, they were relatively insensitive to TSA treatment. Surprisingly, the expression levels of HDAC genes, including the Hdac11, did not increase significantly in the SLO of MOG35-55 immunized mice nor repressed by TSA treatment (Supplementary Fig. 5). These results indicate that MOG35-55 immunization leads to increased transcription of Hdac11 in a CNS-specific fashion, which is rendered sensitive to the action of the histone modifier. These results indicate the possibility that Hdac11 could represent a novel target for the manipulation of PEAE.
Fig. 5 TSA treatment repressed the transcription of Hdac11 selectively in the CNS. 4. Discussion A cardinal assumption has been that the T lymphocytes play a crucial role in the induction and manifestation of multiple sclerosis, and therefore, induction of antigen-specific T cell tolerance is a reasonable approach to treat this debilitating disease. Many attempts, including immunization with various neuronal peptides and T cell vaccinations, have failed to induce T cell tolerance and ameliorate multiple sclerosis symptoms (Wingerchuk and Carter, 2014; Dendrou et al., 2015; Steinman, 2015; Lassmann, 2017). However, the induction of antigen-specific T cell tolerance using immunomodulatory drugs remains unexplored in patients with MS and other autoimmune disorders. Recently, we have demonstrated that treatment with the potent HDAC inhibitor TSA not only reduced the frequencies of Th1, Th17, and Th1/Th17 cells in the SLO and their influx into the CNS but also induced MOG35-55 peptide-specific T cell tolerance (anergy) in NOD mice (Jayaraman et al., 2017). Although anergy was originally reported in a mouse Th1 clone that was suboptimally activated without co-stimulation in vitro (Schwartz et al., 1989; Jayaraman et al., 1992), the underlying mechanisms have not been fully deciphered. Our investigation unraveled a good correlation between TSA-mediated neuroprotection and downregulation of selected immune response-related genes both in the CNS and SLO. Whereas these data may not directly impinge upon histone modifier-facilitated MOG35-55 peptide-specific T cell tolerance, they highlight the possible impact of differential gene expression on PEAE. The epigenetic approach unraveled an inverse relationship between the expression levels of a small set of genes and neuroprotection. In the CNS of PEAE NOD mice, Il17a expression was upregulated for over a month (Fig. 2), unlike its expression at the peak of the monophasic EAE (Kreymborg et al., 2007; Evangelidou et al., 2014; Borjini et al., 2016). Prolonged expression of Nos2 in the CNS (Fig. 2B) is congruent with the association of iNOS-positive macrophages and glial cells in demyelinating pathology (Tran et al., 1997). In addition to the Nos2 (Fig. 3C), the subunits of IL-27, namely IL-27p28, and IL-27EBi3, were prominently upregulated at the mRNA level in the SLO (Fig. 3B-C). This is consistent with an encephalitogenic role of IL-27 suggested by the suppression of the ongoing EAE following administration of the neutralizing antibody against the IL-27p28 subunit (Goldberg et al., 2004). Besides, TSA treatment reduced the steady-state expression of Mif in both the CNS and SLO (Fig. 2E, Fig. 3E), consistent with a proposed pathogenic role of MIF in monophasic EAE (Powell et al., 2005). We have previously shown that TSA treatment diminished the numbers of splenocytes expressing the co-inhibitory ligand CD274 (PD-L1) (Jayaraman et al., 2018). Consistently, the Cd274 mRNA level was also repressed in the CNS (Fig. 2G) and SLO (Fig. 3F) of TSA-treated PEAE mice, suggesting a role for this co-inhibitory ligand in encephalomyelitis, as proposed (Jayaraman et al., 2018). Also, the expression of Ahr, uniquely required for the generation of T cells responsible for the late-onset EAE (Nakahama et al., 2013) was diminished in mice protected by TSA treatment (Fig. 2F). Collectively, the TSA-mediated downregulation of both inducible and constitutively expressed genes appears to be inversely proportional to the severity of the neuronal disease. However, it remains to be determined whether the expression levels of these genes may serve as biomarkers for the diagnosis of MS. Nevertheless, the information uncovered in the NOD mouse model may provide a framework for potential MS treatment, typically diagnosed as clinically isolated syndrome followed by years of asymptomatic period (Dendrou et al., 2015). The upregulation of the transcription of Hdac11 in a CNS-specific fashion in mice immunized with MOG35-55 (Fig. 5) represents the first report of differential expression of Hdac genes in vivo. Although the non-selective HDAC inhibitor TSA did not diminish the HDAC11 enzymatic activity in vitro (Bradner et al., 2010), our data demonstrated the control of Hdac11 by TSA at the transcriptional level. Since Hdac11 gene expression was determined in the SC devoid of peripheral blood contamination, its reduction appears to be a direct effect of TSA on the CNS resident cells. Although we have not identified the cellular source of Hdac11 in the spinal cord, previous work suggested that Hdac11 knockdown increased Il10 expression in peripheral antigen-presenting cells resulting in immunosuppression in vitro (Villagra et al., 2009). In contrast, TSA-induced tolerance in MOG35-55-specific T lymphocytes (Jayaraman et al., 2017) did not accompany the upregulated transcription of Il10 in the CNS or SLO (see Supplementary Fig. 1). Although the deletion of Hdac1 selectively in T cells was reported to prevent EAE (Göschl et al., 2018), immunization with MOG35-55 neither upregulated the expression of this gene, nor the epigenetic drug influenced its transcription in the SLO or CNS (Fig. 5, Supplementary Fig. 5). Thus, the histone hyperacetylating drug appears to primarily target the Hdac11 gene in the CNS of PEAE mice and the elucidation of the genes that are under the control of Hdac11 may provide novel insights into the mechanisms of PEAE. To our surprise, the epigenetic drug treatment failed to validate the purported roles of the genes critical for the development of encephalitogenic Th1 and Th17 subsets, including Ifng, Il12, Il18, Il23, Tbet, Rorgt, Gata3, and Eomes. The disruption of the IFN-γ gene failed to influence EAE development (Ferber et al., 1995), indicating a lack of IFN-γ-expressing cells in neurodegeneration. Consistently, the reduction of clinical symptoms by TSA treatment did not accompany the transcriptional repression of IFN-γ (Supplementary Fig. 1). Besides, the drug-mediated tolerance induction was evident without repressed transcription of Tbet, Il18, and Il12 genes critical for Th1 cell development (O'Connor et al., 2013; Lalor et al., 2011; Gran et al., 2004) (see Supplementary Fig. 3). Moreover, the transcription of Il23 (Kreymborg et al., 2007; El-Behi et al., 2011) and Rorgt (Martinez et al., 2014), respectively, involved in Th17 and Th2 cell generation was also not diminished by TSA treatment. Yet, the histone modifier reduced the overall numbers of Th17 and Th1/Th17 cells in the SLO (Jayaraman et al., 2017). Furthermore, the drug treatment failed to decrease the transcription of Csf2 (see Supplementary Fig. 2) despite the diminished numbers of GM-CSF-co-expressing Th1 and Th17 cells found in the SLO of TSA-treated mice (Jayaraman et al., 2017). These data indicate that T cell tolerance induction by epigenetic modulation of the genome does not involve selective suppression of genes required for the generation of functionally distinct Th cell subsets. Further work is necessary to decipher the underlying mechanisms of T cell tolerance. Another deviation from the conventional idea of immunoregulation is the lack of the modulation of Il10 and Foxp3 both in the CNS and SLO of TSA-treated mice (Supplementary Fig. 4). Although Foxp3+ T regulatory cells are considered critical for immunoregulation (Hori et al., 2003), their role in EAE is less compelling (Jayaraman et al., 2017; Danikowski et al., 2017; Jayaraman and Prabhakar, 2019). Whereas TSA treatment increased Foxp3 expression and promoted T regulatory cell function via the upregulation of Hdac9 in a different experimental model (Tao et al., 2007), our results contradict these findings. It is also noteworthy that TSA treatment downregulated Foxp3 expression and lowered the numbers of CD4+CD25+ T regulatory cells (Liu et al., 2010). Nevertheless, our data indicate that TSA-mediated protection from PEAE in autoimmune-prone NOD mice is independent of IL-10 and Foxp3+ T regulatory cells. Differential RNA display (Van Lint et al., 1996) and microarray analysis of gene expression (Jayaraman et al., 2013) indicated that histone hyperacetylation by TSA treatment could have positive, negative, or no effect on gene transcription. Our qRT-PCR analysis showed that out of 41 genes interrogated, 11 were consistently downregulated over multiple time points following TSA treatment (vide supra). Rarely, TSA treatment can also increase the transcription of genes due to the transcriptional suppression of repressor complexes that control gene expression. Thus, Dec1 was selectively upregulated in the CNS by the histone modifier, suggesting a role in protection against PEAE (Fig. 2H). This is in contrast to the finding that the genetic deletion of Dec1 (Bhlhe40-/-) afforded resistance to EAE induction (Lin et al., 2016). Like an earlier study (Hidaka et al., 2014), we found that the expression of the CCL2 gene was dramatically increased in the SLO of MOG35-55 immunized NOD mice (Fig. 4). However, TSA treatment failed to influence its expression. Moreover, there is a disconnect between the CCL2 mRNA expression in the SLO and the release of the CCL2 protein into the peripheral blood. It has been proposed that CCL2 expressed by the CNS resident astrocytes may serve as a target for MS treatment (Mahad and Ronsohoff, 2003; Moreno et al., 2014). However, Ccl2 and Ccr2 genes are refractory to histone hyperacetylation, and therefore do not appear to be essential for PEAE induction and manifestation. This observation does not come as a surprise since the level of CCL2 was also reported to be lower in relapsing-remitting MS patients (Narikawa et al., 2004; Moreira et al., 2006), and the IFN-beta 1a therapy reduced relapses while increasing the level of CCL2 in MS patients (Szczuciński and Losy, 2004). Thus, these data do not support the hypothesis that the CCL2: CCR2 system may impact neurodegeneration. It is noteworthy that TSA administration induced neuroprotection and gene regulation similarly in prediabetic, 9-13 weeks old, and aged (17-weeks old) mice (Jayaraman et al., 2017; Fig. 1-5). The administration of the epigenetic drug for one to six weeks bestowed comparable neuroprotection and gene regulation irrespective of the age of the mice. Circumstantial evidence also indicates the lack of influence of diabetes on PEAE induction. As expected, the prediabetic (9-13-weeks old) mice were normoglycemic at the time of immunization with MOG35-55 and remained non-diabetic as long as 27 weeks of age. The lack of diabetes in immunized mice could be due to the action of the microbial products present in the complete Freund’s adjuvant. In fact, we have shown that the administration of complete Freund’s adjuvant alone is sufficient to prevent prediabetic NOD mice from developing overt diabetes (Jayaraman and Jayaraman, 2018). Although a fraction of the older (17 weeks old) mice developed diabetes, it did not impede PEAE induction (Fig. 1B-C) nor the modulation of gene expression in response to TSA treatment (Fig. 2-5). Collectively, these data demonstrate that the induction of PEAE and epigenetic regulation of gene expression is unrelated to the age or the glycemic status of mice. Conclusions The analysis of the 41 genes using qRT-PCR unraveled differential regulation of gene expression in the CNS and SLO by an epigenetic drug. This study has highlighted the roles of Il4, Hdac11, Cd274, and Cd39 in the CNS in addition to validating previously implicated genes, Il17a, Il22, iNos, Ahr, and Mif in neuroinflammation. On the other hand, in the SLO, the roles of Il22, Il27, Nos2, and Mif were confirmed while indicating the participation of Cd274 and Cd39 in PEAE. Some of these genes are induced by immunization, while others are constitutively expressed. Surprisingly, many other genes previously implicated in EAE were refractory to histone hyperacetylation mediated transcriptional regulation. Hence, it will be difficult to conclude whether these genes do not contribute to neuroinflammation. Nevertheless, our data suggest that the drug-induced histone hyperacetylation is a promising strategy to treat demyelination and axonal damage by modifying the expression of selected genes. Although the therapeutic potential of HDAC inhibitors for the treatment of multiple sclerosis has been entertained (Faraco et al., 2011), direct evidence is lacking (Göbel et al., 2018). The data generated in the NOD mouse model provide a framework for a similar pharmacological approach to treat MS patients. Author contributions AJ conducted experiments, collected data, edited the graphic and the manuscript. KA, RK, AS, AK, and MK performed experiments, collected data, and approved the manuscript. SJ conceived the project, designed and implemented experiments, analyzed the data, and wrote the paper. Funding This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors. Acknowledgments Mark Holterman and Bellur Prabhakar are acknowledged for the support of this work. References Basso, A.S., Frenkel, D., Quintana, F.J., Costa-Pinto, F.A., Petrovic-Stojkovic, S., Puckett, L., Monsonego, A., Bar-Shir A., Engel Y., Gozin M., Weiner H.L., 2008. Reversal of axonal loss and disability in a mouse model of progressive multiple sclerosis. J. Clin. Invest. 118, 1532-1543. https://doi.org/10.1172/JCI33464. Borjini, N., Fernández, M., Giardino L., Calzà, L., 2016. Cytokine and chemokine alterations in tissue, CSF, and plasma in early presymptomatic phase of experimental allergic encephalomyelitis (EAE), in a rat model of multiple sclerosis. J. Neuroinflammation 13, 291. https://doi.org.10.1186/s12974-016-0757-6. Bradner, J.E., West, N., Grachan, M.L., Greenberg, E.F., Haggarty, S.J., Warnow, T., Mazitschek, R., 2010. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 6, 238-243. https://doi.org.10.1038/nchembio.313. Dang, P.T., Bui, Q., D'Souza, C.S., Orian, J.M., 2015. Modelling MS: chronic-relapsing EAE in the NOD/Lt mouse strain. Curr. Top. Behav. Neurosci. 26, 143-177. https://doi.org.10.1007/7854_2015_378. Danikowski, K.M., Jayaraman, S., Prabhakar, B.S., 2017. Regulatory T cells in multiple sclerosis and myasthenia gravis. J. Neuroinflammation 14, 117. https://doi.org.10.1186/s12974-017-0892-8. Dendrou, C.A., Fugger, L., Friese, M.A., 2015. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 15, 545-558. https://doi.org.10.1038/nri3871. De Ruijter, A.J., van Gennip, A.H., Caron, H.N., Kemp, S., van Kuilenburg, A.B., 2003. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 370, 737-749. PMID:12429021. El-Behi, M., Ciric, B., Dai, H., Yan, Y., Cullimore, M., Safavi, F., Zhang, G.X., Dittel, B.N., Rostami, A., 2011. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol. 12, 568-575. https://doi.org.10.1038/ni.2031. Evangelidou, M., Karamita, M., Vamvakas, S.S., Szymkowski, D.E., Probert, L., 2014. Altered expression of oligodendrocyte and neuronal marker genes predicts the clinical onset of autoimmune encephalomyelitis and indicates the effectiveness of multiple sclerosis-directed therapeutics. J. Immunol. 192, 4122-3133. https://doi.org.10.4049/jimmunol.1300633. Falcone, M., Rajan, A.J., Bloom, B.R., Brosnan, C.F., 1998. A critical role for IL-4 in regulating disease severity in experimental allergic encephalomyelitis as demonstrated in IL-4-deficient C57BL/6 mice and BALB/c mice. J. Immunol. 160, 4822–4830. PMID:9590229. Faraco, G., Cavone L., Chiarugi., 2011. The therapeutic potential of HDAC inhibitors in the treatment of multiple sclerosis. Mol. Med. 17, 442-447. PMID 21373721. Ferber, I.A., Brocke, S., Taylor-Edwards, C., Ridgway, W., Dinisco, C., Steinman, L., Dalton, D., Fathman, C.G., 1996. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J. Immunol. 156, 5-7. PMID:8598493. Fernando, V., Omura, S., Sato, F., Kawai, E., Martinez, N.E., Elliott, S.F., Yoh, K., Takahashi, S., Tsunoda, I., 2014. Regulation of an autoimmune model for multiple sclerosis in Th2-biased GATA3 transgenic mice. Int. J. Mol. Sci. 15, 1700-1708. https://doi.org.10.3390/ijms15021700. Fox, R.J., Chataway, J., 2017. Advancing trial design in progressive multiple sclerosis. Mult. Scler. 23, 1573-1578. https://doi.org.10.1177/1352458517729768 Göbel, K., Ruck, T., Meuth, S.G., 2018. Cytokine signaling in multiple sclerosis: Lost in translation. Mult. Scler. 24, 432-439. https://doi.org.10.1177/1352458518763094. Goldberg, R., Wildbaum, G., Zohar, Y., Maor, G., Karin, N., 2004. Suppression of ongoing experimental autoimmune encephalomyelitis by neutralizing the function of the p28 subunit of IL-27. J. Immunol. 173, 6465-6471. https://doi.org.10.4049/jimmunol.173.10.6465. Göschl, L., Preglej, T., Hamminger, P., Bonelli, M., Andersen, L., Boucheron, N., Gülich, AF., Müller, L., Saferding, V., Mufazalov, IA., Hirahara, K., Seiser, C., Matthias, P., Penz, T., Schuster, M., Bock, C., Waisman, A., Steiner, G., Ellmeier, W., 2018. A T cell-specific deletion of HDAC1 protects against experimental autoimmune encephalomyelitis. J. Autoimmun. 86, 51-61. https://doi.org.10.1016/j.jaut.2017.09.008. Gran, B., Zhang, G.X., Rostami, A., 2004. Role of the IL-12/IL-23 system in the regulation of T-cell responses in central nervous system inflammatory demyelination. Crit. Rev. Immunol. 24, 111-128. PMID:15581393. Hidaka, Y., Inaba, Y., Matsuda, K., Itoh, M., Kaneyama, T., Nakazawa, Y., Koh, C.S., Ichikawa, M., 2014. Cytokine production profiles in chronic relapsing-remitting experimental autoimmune encephalomyelitis: IFN-γ and TNF-α are essential participants in the first attack but not in the relapse. J. Neurol. Sci. 340, 117-122. https://doi.org/10.1016/j.jns.2014.02.039. Hori, S., Nomura, T., Sakaguchi, S., 2003. Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057-1061. https://doi.org.10.1126/science.1079490. Jayaraman, S., Muthukkaruppan, V.R., 1977. In vitro correlate of transplantation immunity: spleen cell migration inhibition in the lizard, Calotes Versicolor. Dev. Comp. Immunol. 1, 133-143. https://doi.org.10.1016/s0145-305x(77)80006-2. Jayaraman, S., Luo, Y., Dorf, M.E., 1992. Tolerance induction in T helper (Th1) cells by thymic macrophages. J. Immunol. 148, 2672-2681. PMID:1533409. Jayaraman, S., Patel, T., Patel, V., Garza, R., Ajani, S., Jayaraman, A., Singh, R., Kwon, S., Rondelli, D., Prabhakar, S., Holterman, M., 2010. Transfusion of nonobese diabetic mice with allogeneic newborn blood ameliorates autoimmune diabetes and modifies the expression of selected immune response genes. J. Immunol. 184, 3008-3015. PMID:20164427. Jayaraman, S., Patel, A., Jayaraman, A., Patel, V., Holterman, M., Prabhakar, B., 2013. Transcriptome analysis of epigenetically modulated genome indicates signature genes in manifestation of type 1 diabetes and its prevention in NOD mice. PLoS One 8, e55074. https://doi.org.10.1371/journal.pone.0055074. Jayaraman, A., Soni, A., Prabhakar, B.S., Holterman, M., Jayaraman, S., 2017. The epigenetic drug Trichostatin A ameliorates experimental autoimmune encephalomyelitis via T cell tolerance induction and impaired influx of T cells into the spinal cord. Neurobiol. Dis. 108, 1-12. https://doi.org.10.1016/j.nbd.2017.07.015. Jayaraman, A., Sharma, M., Prabhakar, B., Holterman, M., Jayaraman, S., 2018. Amelioration of progressive autoimmune encephalomyelitis by epigenetic regulation involves selective repression of mature neutrophils during the preclinical phase. Exp. Neurol. 304, 14-20. https://doi.org.10.1016/j.expneurol.2018.02.008. Jayaraman, S., Jayaraman, A., 2018. Long-term provision of acidified drinking water fails to influence autoimmune diabetes and encephalomyelitis. J. Diab. Res. 2018:3424691. PMID:30035128. Jayaraman, S., Prabhakar, B.S., 2019. Immune tolerance in autoimmune central nervous system disorders. in: Mitoma, H., Manto, M., (Eds.) Autoimmune Central Nervous Disorders. Springer Nature, London, pp.143-166. https://doi.org.10.1007/978-3-030-19515-1_5. Kandagaddala, L.D., Kang, M.J., Chung, B.C., Patterson, T.A., Kwon, O.S., 2012. Expression and activation of matrix metalloproteinase-9 and NADPH oxidase in tissues and plasma of experimental autoimmune encephalomyelitis in mice. Exp. Toxicol. Pathol. 64, 109-114. https://doi.org.10.1016/j.etp.2010.07.002. Kreymborg, K., Etzensperger, R., Dumoutier, L., Haak, S., Rebollo, A., Buch, T., Heppner, F.L., Renauld, J.C., Becher, B., 2007. IL-22 is expressed by Th17 cells in an IL-23-dependent fashion, but not required for the development of autoimmune encephalomyelitis. J. Immunol. 179, 8098-8104. https://doi.org.10.4049/jimmunol.179.12.8098. Lassmann, H., 2017. Targets of therapy in progressive MS. Mult. Scler. 23, 1593-1599. https://doi.org/10.1177/1352458517729455. Lassmann, H., Bradl, M., 2017. Multiple sclerosis: experimental models and reality. Acta Neuropathol. 133, 223-244. https://doi.org/10.1007/s00401-016-1631-4. Lalor, S.J., Dungan, L.S., Sutton, C.E., Basdeo, S.A., Fletcher, J.M., Mills, K.H., 2011. Caspase-1-processed cytokines IL-1beta and IL-18 promote IL-17 production by gammadelta and CD4 T cells that mediate autoimmunity. J. Immunol. 186, 5738-5748. https://doi.org.10.4049/jimmunol.1003597. Li, J., Gran, B., Zhang, G.X., Rostami, A., Kamoun, M., 2005. IL-27 subunits and its receptor (WSX-1) mRNAs are markedly up-regulated in inflammatory cells in the CNS during experimental autoimmune encephalomyelitis. J. Neurol. Sci. 232, 3-9. https://doi.org.10.1016/j.jns.2004.12.013. Lin, C.C., Bradstreet, T.R., Schwarzkopf, E.A., Jarjour, N.N., Chou, C., Archambault, A.S., Sim, J., Zinselmeyer, B.H., Carrero, J.A., Wu, G.F., Taneja, R., Artyomov, M.N., Russell, J.H., Edelson, B.T., 2016. IL-1-induced Bhlhe40 identifies pathogenic T helper cells in a model of autoimmune neuroinflammation. J. Exp. Med. 213, 251-271. https://doi.org.10.1084/jem.20150568. Liu, Z., Zhang, C., Sun, J., 2010. Deacetylase inhibitor trichostatin A down-regulates Foxp3 expression and reduces CD4+CD25+ regulatory T cells. Biochem. Biophys. Res. Commun. 400, 409-412. https://doi.org.10.1016/j.bbrc.2010.08.090. Mahad, D.J., Ransohoff, R.M., 2003. The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Semin. Immunol. 15, 23-32. PMID:12495638. Martinez, N.E., Sato, F., Omura, S., Kawai, E., Takahashi, S., Yoh, K., Tsunoda, I., 2014. RORγt, but not T-bet, overexpression exacerbates an autoimmune model for multiple sclerosis. J. Neuroimmunol. 276, 142-149. https://doi.org.10.1016/j.jneuroim.2014.09.006. Mascanfroni, I.D., Yeste, A., Vieira, S.M., Burns, E.J., Patel, B., Sloma, I., Wu, Y., Mayo, L., Ben-Hamo, R., Efroni, S., Kuchroo, V.K., Robson, S.C., Quintana, F.J., 2013. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat. Immunol. 14,1054-1063. https://doi.org.10.1038/ni.2695. McQualter, J.L., Darwiche, R., Ewing, C., Onuki, M., Kay, T.W., Hamilton, J.A, Reid, H.H., Bernard, C.C., 2001. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J. Exp. Med. 194, 873-882. https://doi.org:10.1084/jem.194.7.873. Moreira, M.A., Tilbery, C.P., Monteiro, L.P., Teixeira, M.M., Teixeira, A.L., 2006. Effect of the treatment with methylprednisolone on the cerebrospinal fluid and serum levels of CCL2 and CXCL10 chemokines in patients with active multiple sclerosis. Acta Neurol. Scand. 114, 109-113. https://doi.org/10.1111/j.1600-0404.2006.00629. Moreno, M., Bannerman, P., Ma, J., Guo, F., Miers, L., Soulika, A.M., Pleasure, D., 2014. Conditional ablation of astroglial CCL2 suppresses CNS accumulation of M1 macrophages and preserves axons in mice with MOG peptide EAE. J. Neurosci. 34, 8175-8185. https://doi.org.10.1523/JNEUROSCI.1137-14.2014. Nakahama, T., Hanieh, H., Nguyen, N.T., Chinen, I., Ripley, B., Millrine, D., Lee, S., Nyati, K.K., Dubey, P.K., Chowdhury, K., Kawahara, Y., Kishimoto, T., 2013. Aryl hydrocarbon receptor-mediated induction of the microRNA-132/212 cluster promotes interleukin-17-producing T-helper cell differentiation. Proc. Natl. Acad. Sci. USA 110, 11964-11969. https://doi.org.10.1073/pnas.1311087110. Narikawa, K., Misu, T., Fujihara, K., Nakashima, I., Sato, S., Itoyama, Y., 2004. CSF chemokine levels in relapsing neuromyelitis optica and multiple sclerosis. J. Neuroimmunol. 149, 182-186. https://doi.org/ 10.1016/j.jneuroim.2003.12.010. Niino, M., Ogata, A., Kikuchi, S., Tashiro, K., Nishihira, J., 2000. Macrophage migration inhibitory factor in the cerebrospinal fluid of patients with conventional and optic-spinal forms of multiple sclerosis and neuro-Behçet's disease. J. Neurol. Sci. 179, 127-131. https://doi.org. 10.1016/s0022-510x(00)00397-x. O'Connor, R.A., Cambrook, H., Huettner, K., Anderton, S.M., 2013. T-bet is essential for Th1-mediated, but not Th17-mediated, CNS autoimmune disease. Eur. J. Immunol. 43, 2818-2823. https://doi.org.10.1002/eji.201343689. Okuda, Y., Sakoda, S., Yanagihara, T., 1998. The pattern of cytokine gene expression in lymphoid organs and peripheral blood mononuclear cells of mice with experimental allergic encephalomyelitis. J. Neuroimmunol. 87, 147-155. PMID:9670856. Patel, T., Patel, V., Singh, R., Jayaraman, S., 2011. Chromatin remodeling resets the immune system to protect against autoimmune diabetes in mice. Immunol. Cell Biol. 89:640-649. PMID:21321581. Pierson, E.R., Goverman, J.M., 2017. GM-CSF is not essential for experimental autoimmune encephalomyelitis but promotes brain-targeted disease. JCI Insight 2, e92362. https://doi.org.10.1172/jci.insight.92362. Ponomarev, E.D., Maresz, K., Tan, Y., Dittel, B.N., 2007. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J. Neurosci. 27, 10714-10721. https://doi.org.10.1523/JNEUROSCI.1922-07.2007. Powell, N.D., Papenfuss, T.L., McClain, M.A., Gienapp, I.E, Shawler, T.M., Satoskar, A.R., Whitacre, C.C., 2005. Cutting edge: macrophage migration inhibitory factor is necessary for progression of experimental autoimmune encephalomyelitis. J. Immunol. 175, 5611-5614. https://doi.org.10.4049/jimmunol.175.9.5611. Raveney, B.J., Oki, S., Hohjoh, H., Nakamura, M., Sato, W., Murata, M., Yamamura, T., 2015. Eomesodermin-expressing T-helper cells are essential for chronic neuroinflammation. Nat. Commun. 6, 8437. https://doi.org.10.1038/ncomms9437. Rempe, R.G., Hartz, A.M, Bauer, B., 2016. Matrix metalloproteinases in the brain and blood-brain barrier: Versatile breakers and markers. J. Cereb. Blood Flow Metab. 36, 1481-1507. https://doi.org.10.1177/0271678X16655551. Rivers, T.M., Sprunt, D.H, Berry, G.P., 1933. Observations on attempts to produce acute disseminated encephalomyelitis in monkeys. J. Exp. Med. 58, 39-53. https://doi.org.10.1084/jem.58.1.39. Schwartz, RH., Mueller, DL., Jenkins, MK., Quill, H., 1989. T-cell clonal anergy. Cold Spring Harb. Symp. Quant. Biol. 54, 605-610. PMID 2534840. Slavin, A., Ewing, C., Liu, J., Ichikawa M., Slavin J., Bernanrd, C.C., 1998. Induction of a multiple sclerosis-like disease in mice with an immunodominant epitope of myelin oligodendrocyte glycoprotein. Autoimmunity 28, 109-120. https://doi.org.10.3109/08916939809003872. Steinman, L., 2015. The re-emergence of antigen-specific tolerance as a potential therapy for MS. Mult. Scler. 21, 1223-1238. https://doi.org.10.1177/1352458515581441. Szczuciński, A., Losy, J., 2004. Long-term effect of IFN-beta 1a therapy on CCL2 (MCP-1) chemokine in patients with multiple sclerosis. Folia Neuropathol. 42, 15-18. PMID:15119740. Tao, R., de Zoeten, E.F., Ozkaynak, E., Chen, C., Wang, L., Porrett, P.M., Li, B., Turka, L.A., Olson, E.N., Greene, M.I., Wells, A.D., Hancock, W.W., 2007. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat. Med. 13, 1299-1307. https://doi.org.10.1038/nm1652. Tintore, M., Vidal-Jordana, A., Sastre-Garriga, J., 2019. Treatment of multiple sclerosis - success from bench to bedside. Nat. Rev. Neurol. 15, 53-58. https://doi.org/10.1038/s41582-018-0082-z. Tran, E.H., Hardin-Pouzet, H., Verge, G., Owens, T., 1997. Astrocytes and microglia express inducible nitric oxide synthase in mice with experimental allergic encephalomyelitis. J. Neuroimmunol. 74, 121-129. https://doi.org.10.1016/s0165-5728(96)00215-9. Van Lint, C., Emiliani, S., Verdin, E., 1996. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr. 5, 245-253. PMID:8723390. Villagra, A., Cheng, F., Wang, H.W., Suarez, I., Glozak, M., Maurin, M., Nguyen, D., Wright, K.L., Atadja, P.W., Bhalla, K., Pinilla-Ibarz, J., Seto, E., Sotomayor, E.M., 2009. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 10, 92-100. https://doi.org.10.1038/ni.1673. Wingerchuk, D.M., Carter, J.L., 2014. Multiple sclerosis: current and emerging disease-modifying therapies and treatment strategies. Mayo Clin. Proc. 89, 225-240. https://doi.org.10.1016/j.mayocp.2013.11.002.
Copyright: © 2020 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |