|
|
|
Free Neuropathology 1:17 (2020) |
|
Review |
|
Multiple system atrophy - a clinicopathological update |
|
Kurt A. Jellinger |
|
Institute of Clinical Neurobiology, Vienna, Austria |
|
Corresponding author: |
|
Submitted: 25 May 2020 Accepted: 24 June 2020 Copyedited by: Nicole Schwab Published: 3 July 2020 |
|
Keywords: Multiple system atrophy, α-synuclein, Glio-neuronal degeneration, Animal models, Etiopathogenesis, Prion-like seeding, Biomarkers, Experimental therapeutics |
|
Abstract Multiple system atrophy (MSA) is a fatal, adult-onset neurodegenerative disorder of uncertain etiology, clinically characterized by various combinations of Levo-dopa-unresponsive parkinsonism, and cerebellar, motor, and autonomic dysfunctions. MSA is an α-synucleinopathy with specific glioneuronal degeneration involving striatonigral, olivopontocerebellar, autonomic and peripheral nervous systems. The pathologic hallmark of this unique proteinopathy is the deposition of aberrant α-synuclein (αSyn) in both glia (mainly oligodendroglia) and neurons forming pathological inclusions that cause cell dysfunction and demise. The major variants are striatonigral degeneration (MSA with predominant parkinsonism / MSA-P) and olivopontocerebellar atrophy (MSA with prominent cerebellar ataxia / MSA-C). However, the clinical and pathological features of MSA are broader than previously considered. Studies in various mouse models and human patients have helped to better understand the molecular mechanisms that underlie the progression of the disease. The pathogenesis of MSA is characterized by propagation of disease-specific strains of αSyn from neurons to oligodendroglia and cell-to-cell spreading in a "prion-like" manner, oxidative stress, proteasomal and mitochondrial dysfunctions, myelin dysregulation, neuroinflammation, decreased neurotrophic factors, and energy failure. The combination of these mechanisms results in neurodegeneration with widespread demyelination and a multisystem involvement that is specific for MSA. Clinical diagnostic accuracy and differential diagnosis of MSA have improved by using combined biomarkers. Cognitive impairment, which has been a non-supporting feature of MSA, is not uncommon, while severe dementia is rare. Despite several pharmacological approaches in MSA models, no effective disease-modifying therapeutic strategies are currently available, although many clinical trials targeting disease modification, including immunotherapy and combined approaches, are under way. Multidisciplinary research to elucidate the genetic and molecular background of the noxious processes as the basis for development of an effective treatment of the hitherto incurable disorder are urgently needed. Abbreviations αSyn - α-synuclein, ADNC - Alzheimer disease neuropathological changes, BG - basal ganglia, CAA - cerebral amyloid angiopathy, CI - cognitive im-pairment, CN - caudate nucleus, CNS - central nervous system, CSF - cerebrospinal fluid, DAT - dopamine transporter, FTLD - frontotemporal lobar degeneration GCI - glial cytoplasmic inclu-sion, GDNF - glia-derived neurotrophic factors, GP - globus pallidus, GWAS - genome-wide association study, HPR - hyperintensive putaminal rim, LBD - Lewy body disease, LBs - Lewy bodies, MBP - myelin basic protein, MCI - mild cognitive impairment, MSA - multiple system atrophy, MSA-C - MSA with prominent cerebellar ataxia, MSA-P - MSA with predominant parkinsonism, OPC - olivopontocerebellar, OPCA - olivo¬pontocerebellar atrophy, OS - oxidative stress, PART - primary age-related tauopathy, PD - Parkinson disease, PET - positron emission tomography, PrPC - cellular prion protein, PSP - progressive supranuclear palsy, SN - substantia nigra, SND - striatonigral degeneration, tg - transgenic, TPPP - tubulin polymerization-promoting protein, wt - wild type Introduction Multiple system atrophy (MSA) is a rare adult-onset and lethal neurodegenerative disorder clinically characterized by rapidly progressing autonomic and motor dysfunctions. The pathological hallmark of MSA, a specific form of α-synucleinopathy, is abnormal accumulation of fibrillar α-synuclein (αSyn) in oligodendrocytes as glial cytoplasmic inclusions (GCI) [1], which may represent a primary pathologic event [2]. Degeneration of many neuronal pathways causes multifaceted clinical phenotypes: a parkinsonian variant (MSA-P), associated with striatonigral degeneration (SND), and a cerebellar (MSA-C) variant with olivopontocerebellar atrophy (OPCA) [3]. In addition to combined or "mixed" MSA, there are several disease variants [4-6]. The underlying molecular mechanisms are poorly understood, but converging evidence suggests that a "prion-like" spreading of disease-specific αSyn strains is involved in the pathogenic cascade leading to a specific multisystem neurodegeneration in this (oligodendro)glioneuronal proteinopathy [4, 7-12]. The aim of the present review is to describe recent advances in MSA neuropathology, clinical diagnosis, neuroimaging, and candidate biomarkers. It further provides an overview of the mechanisms underlying MSA pathogenesis and of possible novel therapeutic targets that have emerged from animal studies and preclinical interventional trials [13-16]. Epidemiology MSA is a rare disease with an estimated incidence of 0.6-0.7/100,000 person-years [17], although studies from Russia and Northern Sweden have reported incidences of 0.1 and 2.4/100,000 person-years, respectively [18, 19]. Prevalence estimates range from 1.9 to 4.9/100,000 [20] but may reach up to 7.8 after the age of 40 years [21], and up to 12/100,000 after the age of 70 [22]. MSA-P accounts for 70-80% of cases in the western world [23], whereas MSA-C is more frequent in Asian populations (67-84%) with rather frequent mixed phenotypes [24-27], probably due to genetic and environmental factors [5]. Etiology and genetics MSA is generally considered a sporadic disease [17], but MSA pedigrees with both autosomal dominant and autosomal recessive inheritance patterns have been reported in Europe and Asia [28-33]. A genome-wide association study (GWAS) found an estimated heritability of 2-7% [34], but unlike Parkinson disease (PD), no single gene mutations linked to familial forms and no definite environmental risk factors have been identified [35]. Screening for PD causal genes (MAPT, PDYN, Parkin, PINK1, and several single nucleotide polymorphisms/SNPs) did not reveal any association with MSA [36-38], while LRRK2 exon variants may contribute to its susceptibility [39]. Glucocerebrosidase (GBA) variants were associated with autopsy-proven MSA [40, 41], particularly with MSA-C [42], while others have found no association [43]. Furthermore, C9ORF72 repeat expansions [44] and SNCA polymorphisms as risk factors of MSA [45, 46] have not been confirmed [47-49]. No significant associations of the APOE locus nor the prion PRNP with risk of MSA was observed [50, 51], and there is no evidence of autosomal dominant MSA or of de novo mutations in this disorder [52]. A British family with SNCA mutation showed neuropathologic features of both PD and MSA [53], but they are distinct from PD patients carrying the H50Q or SNCA duplication [54]. None of the nucleotide polymorphisms (FBXO47, ELOVL7, EDN1, etc.) reached genome-wide significance [34], and polymorphisms of the LINGO1 and LINGO2 (nogo receptor interacting protein-1 and -2) do not decrease the risk of MSA [55]. The possible involvement of the SNCA, COQ2, MAPT, GBA1, LRRK2 and C9ORF72 genes in MSA pathogenesis was examined recently [56]. The link between V393A mutations and the COQ2 gene, encoding the coenzyme Q10 (COQ10), and familial or sporadic MSA in Japanese and other Asian populations [44, 57-61] has not been confirmed in other populations [34, 62-64]. Thus, COQ2 polymorphisms may be region-specific and may not represent common genetic factors for MSA. Decreased levels of COQ10 in cerebellum and plasma of MSA patients [65, 66] suggest that its deficiency may contribute to pathogenesis due to decreased electron transport in the mitochondria and increased vulnerability to oxidative stress (OS) [67]. RNA analyses of MSA brain tissue revealed alterations in α- and β-immunoglobuline [68], dysregulations of microRNAs that regulate gene expression in the pons and cerebellum [69, 70], and disruption of long intervening non-coding RNAs (lincRNA) in the frontal cortex along with protein coding genes related to iron metabolism and immune response regulation [71, 72]. Epidemiological studies suggested that epigenetic factors or environmental toxins may be associated with the risk for MSA [73], but there are no convincing data correlating increased risk of MSA with exposure to pesticides, solvents, other toxins, or alcohol consumption [74, 75]. Pathogenesis Although our understanding of MSA remains incomplete, evidence from animal models and human post mortem studies indicates that the accumulation of misfolded αSyn, particularly in oligodendrocytes but also in neurons, plays an essential role in the disease process [10, 76-78]. The impact of the neuronal endosomal-lysosomal system in the processing of αSyn in PD is well established, while lysosomes contribute to the pathogenesis of MSA, enabling oligodendroglial and neuronal uptakt of αSyn [79]. Reduced oligodendrocyte-derived enriched microvesicles (OEMVs) could be an important mechanism related to pathological αSyn aggregation in oligodendrocytes [80]. Although it has been speculated that primary neuronal pathology leads to secondary oligodendroglial degeneration, as suggested by the widespread occurrence of NCIs even in areas lacking GCIs [77], the distribution and severity of neurodegeneration reflects subregional GCI density and supports the assumption that MSA is a primary oligodendrogliopathy [2, 81]. The role of oligodendroglia in introducing the neurodegenerative process was confirmed experimentally in transgenic (tg) mice overexpressing αSyn in oligodendrocytes [10, 13, 82-84]. These and other results highlight the role of endogenous αSyn and p25α in the formation of αSyn assemblies in oligodendrocytes and provide in vivo evidence of the role of oligodendroglial αSyn in the establishment of αSyn pathology in MSA [85]. Early events are an ectopic appearance of αSyn in oligodendrocytes, loss of the cAMP-regulated phosphoprotein of 32kDA (DARPP-32), and calbindin indicating calcium toxicity and disturbance of phosphorylated proteins [86]. Recent findings suggest the possibility of endogenous αSyn accumulation in oligodendrocyte precursor cells that contribute to GCI formation and perturbation of neuronal/glia support in MSA brain [86a]. Reduced OEMVs could be an important mechanism related to pathological αSyn aggregates in oligodendroglia, inducing dysfunction of the SNARE protein complex, which regulates membran fusion in eukaryotic cells. The concentrations of OEMVs in MSA were significantly reduced compared to those in PD [80]. Decreased expression of glia-derived neurotrophic factors (GDNF) in MSA brains [87] indicates that αSyn aggregation in oligodendrocytes impacts their trophic transport to neurons. Oligodendroglial changes are more widespread than αSyn positive GCIs, suggesting that oligodendroglial pathology induces degeneration of the oligodendroglia-myelin-axon-neuron complex [2, 26]. The selectivity of the neurodegeneration in MSA is determined by the interaction of multiple noxious factors. Some of these factors include: ectopic αSyn accumulation in oligodendrocytes and neurons, "prion-like" propagation of disease-specific strains of misfolded αSyn [88], targeting distinct brain regions and cell types [89, 90], impaired protein degradation, proteasomal and mitochondrial dysfunctions [91, 92], alterations of the autophagic pathway [91, 93, 94], perturbed iron homeostasis [95], lipid dysfunction involved in myelin synthesis [96-98], genetic polymorphism [55], microglial activation [97, 99], neuroinflammation [100], proteolytic disturbance, autophagy [101], and microRNA dysregulation [102] driving inflammation, disrupting myelin, and contributing αSyn accumulation via the dysregulation of autophagy and prion mechanisms [103]. These and other factors are contributing to OS, which is suggested to be a major pathogenic factor in MSA and related diseases [104]. These multiple mechanisms interact to result in the system-specific pattern of neurodegeneration in MSA (Fig. 1). TNFα-dependent neuroinflammation may play a key role in MSA pathogenesis, and its relevance has been underlined in various models of the disease [105]. αSyn, which shows specific conformational strains [88, 106] that are primarily generated by neurons, can be toxic once released to the extracellular environment [107] and can spread throughout the brain in a "prion-like" manner [9, 108-111]. Extracellular αSyn, interacting with neuronal and non-neuronal cell types, mediates neuroinflammation and cell-to-cell spread [112, 113]. Neuron-to-oligodendrocyte transport of misfolded αSyn plays a major role in the pathogenesis of MSA [114, 115]. MSA and PD show different phosphorylation signatures of αSyn and distinct seed characteristics, indicating that distinct strains underlie these diseases [90, 116, 117]. After propagation in TgM83 tg mice, strain-specific phenotypic differences are maintained after serial transmission, providing evidence that disease heterogeneity among the synucleinopathies is caused by distinct αSyn strains [89]. MSA strains show several similarities to PD strains, and less so with DLB strains, but more potently induce motor deficits, nigrostriatal degeneration, αSyn spreading, and inflammation, reflecting the aggressive nature of this disease [118].
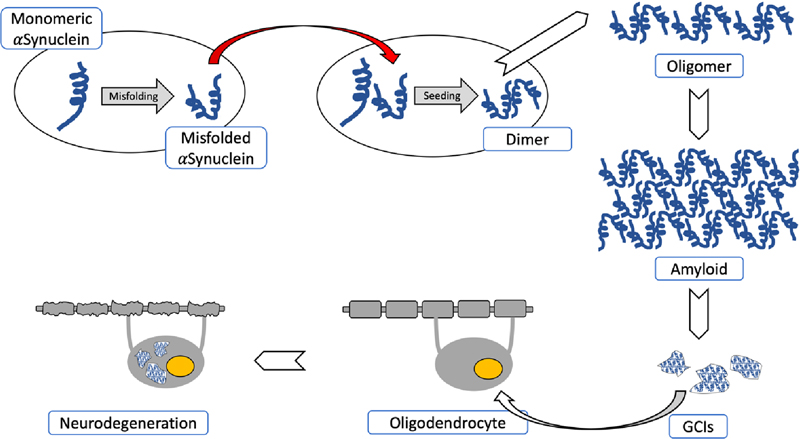
Fig. 1. Pathogenetical features of MSA causing neurodegeneration. Spontaneous misfolding of αSyn results in formation of abnormally folded dimers and further assembly results in oligomers and amyloid formations. αSyn-rich GCIs involving oligodendroglia result in demyelization and neurodegeneration. The red arrow shows the “prion-like” cell-to-cell transfer of misfolded αSyn. Recent animal model studies that only partially replicate the human disorder have provided some progress in our understanding of MSA pathogenesis [13, 15, 84, 119, 120]. Early accumulation of p25α (TPPP), a potent stimulator of αSyn aggregation, may decrease myelin basic protein (MBP), favoring both the deposition and fibrillation of αSyn and changing myelin metabolism [121]. Relocation of p25α from the myelin sheaths to the oligodendroglial soma, due to mitochondrial dysfunction, and the formation of cytoplasmic p25α inclusions precedes the aggregation of transformed αSyn in oligodendrocytes. Endogenous αSyn and p25α induce the formation of pathological αSyn assemblies in oligodendrocytes and provide in vivo evidence of their contribution to the pathogenesis of MSA [85]. Although large inclusions appear in a later disease states, small, soluble assemblies of αSyn, promoted by p25α, are pathogenic [122]. The source of αSyn in oligodendroglia is unclear, but it contains αSyn mRNA expression and αSyn may be secreted by neurons and taken up by oligodendrocytes, which is facilitated by protein Cx32 via direct protein-protein interaction in both neurons and oligodendroglia [115]. 21% of proteins found consistently in GCIs and LBs are synaptic vesicle-related, suggesting that misfolded αSyn may be targeted via vesicle-mediated transport, and may explain the presence of this neuronal protein within GCIs [123]. Thus, MSA represents a specific form of oligodendroglial proteinopathies [124], while others suggest that it is a primary neuronal disease with secondary accumulation of αSyn in oligodendrocytes [77]. Induced pluripotent stem cell (iPSC) studies indicate a pathological phenotype of MSA neurons, independently from oligodendrocytes. These data together with findings in animal models suggest that both neurons and oligodendrocytes are affected in MSA [91]. The disease is currently viewed as a primary synucleinopathy with specific glio-neuronal degeneration developing via the oligo-myelin-axon-neuron complex [2, 4]. Histopathology and molecular pathology The histological core features of MSA encompass the following types of different severity: (1)specific αSyn-immunoreactive inclusion pathology with five types of inclusions: GCIs within oligodendrocytes (also referred to as Papp-Lantos bodies [125], the presence of which is mandatory for the post mortem diagnosis of definite MSA [1]) and less frequently glial nuclear inclusions, neuronal nuclear inclusions, astroglial cytoplasmic inclusions, and neuronal threads, also composed of αSyn [126]; (2) selective neuronal loss and axonal degeneration involving multiple regions of the nervous system; (3) extensive myelin degeneration with pallor and reduction in MBP with astrogliosis; and (4) widespread microglial activation [127] and neuroinflammation [128, 129], with extensive CD4 and CD8 T-cell infiltration [130]. GCIs and the resulting neurodegeneration show a characteristic distribution, involving not only the striatonigral and OPC systems, but also cortical regions, autonomic and motor nuclei in the brainstem, spinal cord, preganglionic autonomic nerve structures [131-134], and the peripheral nervous system [135-138], characterizing MSA as a multi-system/-organ disorder [2, 77, 139]. Phosphorylated αSyn is accumulated in subpial and periventricular astrocytes after long disease duration [140]. However, αSyn-positive astrocytes in subpial and perivascular regions are seen in both MSA and Lewy body disease (LBD), suggesting that this pathology is not a specific feature of MSA [141]. Inclusion pathology GCIs are argyrophilic, triangular, sickle-/half moon-shaped or oval cytoplasmic aggregations, composed of fibrillar αSyn, ubiquitin and various multifunctional proteins, including 14-3-3 protein, LRRK2, aggressomal proteins, etc. [125] (Fig. 2). They form a meshwork of loosely packed filaments or tubules 15-30 nm in diameter with a periodicity of 70-90 nm and straight filaments, both composed of polymerized αSyn granular material and other filaments. The central core contains phosphorylated (ser129) αSyn Cryo-EM showed that αSyn inclusions from MSA are made of two types of filaments, each of which consists of two different protofilaments. Each type contains non-proteinaceous molecules at the interface of the two proteofilaments. Thus, they differ from those in DLB brain, which suggests that distinct conformations/strains are characteristic for specific synucleinopathies. In addition, αSyn filament extracts from MSA tissue differ from those formed in vitro using recombinant proteins, which may have implications for the mechanisms of protein aggregation and neurodegeneration [142]. Soluble αSyn in GCIs differs from the insoluble form in Lewy bodies (LBs) [143]. Purification of αSyn containing GCIs revealed 11.9% αSyn, 2.8% α-β-crystallin, and 1.7% 14-3-3 protein compared to 8.5%, 2.0% and 1.5% in LBs [144]. In the MSA brain, αSyn 140 and 122 isoform levels are increased, whereas αSyn 126 is decreased, in the substantia nigra (SN), striatum, and cerebellum. In early disease states, diffuse αSyn staining in neuronal nuclei and cytoplasm occurs in many gray matter areas, indicating that aggregation of non-fibrillary αSyn occurs early in neurons [26]. Recent studies using a proximity ligation assay revealed a wide distribution of αSyn oligomers not only in oligodendrocytes but also in neocortical neurons and Purkinje cells, suggesting that αSyn oligomer-mediated toxicity is an early event in MSA, inducing neuronal loss in MSA [145].
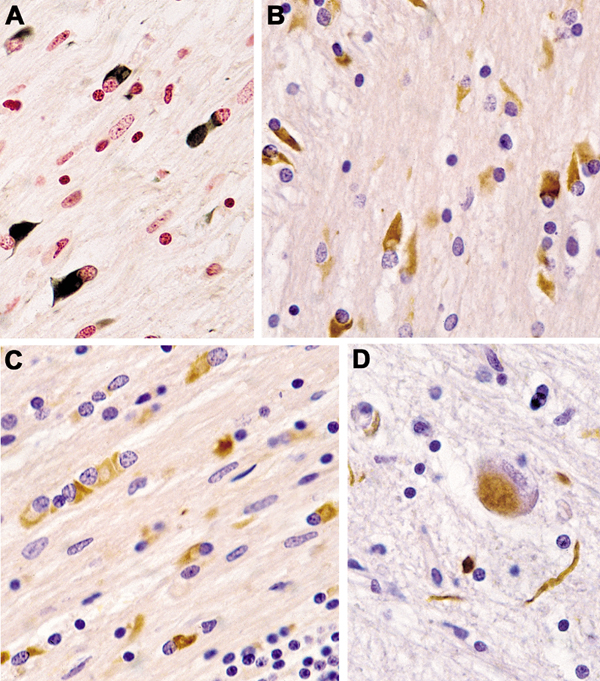
Fig. 2. (A–C) Glial cytoplamic inclusions in MSA: (A) in globus pallidus (Gallyas silver impregnation), (B) in pontine basis (α-Synuclein) and (C) in frontal white matter, anti-ubiquitin. (D) Neuronal cytoplasmic inclusion and neurites in pontine basis (α-Synuclein). (A–D) original magnification 34,000. On the other side, interactions exist between extracellular αSyn and each of the major central nervous system (CNS) cell types. This has thepotential to contribute to secondary disease processes such as neuroinflammation, synaptic dysfunc-tion, and cell-to-cell spread, with vehicles such as microglia and exosomes that mediate spread of αSyn pathology to peripheral brain regions [113]. Cathepsin-D, calpain-1 and kallikrein-6 are elevated in the putamen, pontine basis, and cerebellar white matter, indicating that αSyn accumulation is not due to reduced activity of these proteases, but rather that their upregulation is compensatory to increased αSyn [146]. Iron levels in basal ganglia (BG) and SN are higher in MSA than in PD and controls, indicating perturbed iron homeostasis as a potential pathogenic factor in MSA neurodegeneration [95]. Quantitative analyses of neuronal death and GCI density showed a positive correlation with each other, indicating the pivotal role of GCIs in neuronal death [81, 147], and additionally, both lesions increase with disease duration [148-150]. In the SN, severe neuronal loss is accompanied by low GCI density, indicating that this and other areas affected in early disease have been burned out [139]. Glial nuclear inclusions show a distinct distribution from GCIs (Fig. 2D), and similarly the density of neuronal cytoplasmic inclusions (NCIs) and neuronal nuclear inclusions are unrelated to that of GCIs [151]. NCIs are more widespread and show a hierarchical pattern related to the duration of disease but are independent of neuronal destruction, suggesting that other factors may induce the subtype-dependent neuronal loss [77]. Region-specific astrogliosis is positively correlated with αSyn pathology in MSA, in contrast to PD [152], and in general parallels the severity of neurodegeneration [148]. Microglial activation in degenerated regions accompanies GCI pathology and is most abundant in white matter areas with mild to moderate demyelination [153]. In MSA-C, the cerebellar subcortical white matter and cerebellar brainstem projections are the earliest involved, followed by other CNS regions. Distribution of lesions A grading system for SND was proposed based on semiquantitative assessment of atrophy, neuronal loss, and the presence of GCIs [154]: Neuronal loss in the SN pars compacta is grade 1; extension to the putamen is grade 2; further involvement of the caudate and globus pallidus (GP) is grade 3. Subsequently, the grading system was extended for both SND and OPCA [155]. Of 42 patients, 22 were assigned as MSA-P and 20 as MSA-C, but none displayed "pure" OPCA pathology or more severe OPCA pathology than SND (i.e., OPCA III+SND I/II). These clinicopathological subtypes correlated with initial symptoms and clinical features of both types. Post mortem MRI changes in the putamen (type 1, mild atrophy and isointensity; type 2, atrophy and diffuse hypointensity with a hyperintensive putaminal rim/HPR; type 3, putaminal atrophy and iso- or hypointensity with HPR) reflect various degrees of brain damage [156]. In two large series from the UK and Japan, another grading system for MSA was proposed [148]: each case of SND and OPCA was divided into three grades based on semiquantitative assessment of neuronal loss in regions of interest: for SND, the putamen, GP and SN; and for OPCA the pontine nucleus, cerebellar hemisphere and vermis, inferior olivary nucleus and SN. This classification showed significant clinicopathological correlations. SND phenotypes showed more severe bradykinesia, and the OPCA phenotype more frequently showed cerebellar signs. No patients showed "pure" SND or "pure" OPCA. However, there is an increasing overlap of αSyn pathology with increased duration of the disease the extent of αSyn pathology [157]. Damage to the striatonigral system is most severe in the dorsolateral caudal putamen and lateral SN, suggesting transsynaptic degeneration of the striatonigral fibers. Consistently and severely affected areas are the putamen, CN, SN, pontine and medullary tegmental nuclei, inferior olives, and cerebellar white matter; moderately affected areas are the motor cortex and GP, and mild lesions involve the cingular cortex, hypothalamus, nucleus basalis of Meynert, thalamus, subthalamus, and pontine tegmentum [158]. Degeneration of the GP and SN leads to dysfunction of these inhibitory nuclei projecting to the motor thalamus, but the SN loss is of dopamine, not GABA (gamma aminobutyric acid), neurons. Stereological studies of the BG revealed a substantial loss of neurons in the SN, putamen, and GP, whereas astrocytes were more frequent in the putamen and caudate nucleus (CN). Microglia were found in all CNS regions with greatest frequency in the, otherwise unaffected, red nucleus. These data support the region-specific pattern of pathological changes in MSA [159]. Another neuropathological study showed that the striatonigral region was most severely affected in 34% of SND and in 17% in OPCA cases, while in almost half of them both regions were equally affected [133]. In view of the frequent overlap and mixed forms, the value of grading systems for evaluation of MSA is under discussion [139]. There is widespread involvement of the neocortex with significant loss of neurons and increase of astrocytes and microglia in the frontal and parietal areas, but no change in the total number of oligodendrocytes [160]. Early degeneration of the BG drives late onset cortical atrophy due to fronto-striatal degeneration [161, 162]. Reduced neuronal numbers in the anterior olfactory nucleus and intrabulbar part of the primary olfactory (pyriform) cortex may underlie olfactory dysfunction in MSA [163]. Limbic TDP-43 pathology is rare in MSA, but co-localization with αSyn suggests an interaction between the two molecules [164-167]; TDP-43 positive cases showed significantly older age at death than negative ones, suggesting that TDP-43 pathology in MSA is an age-related phenomenon rather than a disease-specific change [141]. Demyelination of variable intensity affecting all parts of the nervous system [168] is associated with reduction of MBP by about 50% [96]. GCIs and microglial burden are greatest in mild to moderate white matter lesions and decrease with progression of myelin damage that increases with disease duration [169]. The regional vulnerability of the white matter to MSA pathology is poorly understood, but recent GWASs revealed dysregulation of various methylated loci, including HIP1, LMAN2, MOBP, and others, giving the first evidence that DNA methylation changes contribute to the molecular processes altered in MSA [170]. Early MSA stages show increased microglia (about 100%) in the white matter [127], without concomitant astrogliosis or oligodendroglial degeneration [171]. Both microglial activation and αSyn-containing oligodendrocytes trigger neuroinflammation in the white matter [128]. The loss of tubulin polymerization-promoting protein (TPPP)/p25α immunoreactivity correlated significantly with the degree of microglial reaction and loss of MBP density as a marker of tract degeneration [124]. White matter degeneration causes degeneration of neuronal loops, leading to dysfunction of cerebral autoregulation [172]. Gliosis in the degenerated areas of the MSA brain usually correlates with αSyn pathology and the severity of neurodegeneration [153, 173], which is in contrast to PD [174]. Significant increase of monoaminoxidase B (MAO-B), a biomarker of astrogliosis, in the degenerated putamen (+83%) was associated with astrogliosis and showed a positive correlation with αSyn accumulation [175]. Microglial activation accompanying αSyn pathology and phagocytosing degenerating myelin is prominent in all degenerating regions [176], particularly in white matter input tracts to the extrapyramidal system and cerebellum [177]. Stereological studies revealed a significant increase of microglia in the white matter without concomitant astrogliosis and with absence of significant oligodendroglial degeneration [171], suggesting that microglia cells play an important role in the initiation and progression of neurodegeneration in MSA [100, 178]. This is supported by tg mouse models indicating an active contribution of microglial activation by triggering neuroinflammatory responses in the MSA brain [179]. In MSA-C, GCIs are most prominent in the cerebellum, pons, and medulla [169]. The cerebellar Purkinje cells are more severely affected in the vermis, with atrophy of olivary nucleus, cerebellopontine fibers, and pontine basis, causing interruption of specific cerebellocortical circuits [180]. The motor subnetwork in MSA-C is significantly altered in both BG and cerebellar connectivity [181], with hyperintensity of the middle cerebellar peduncle [182]. Involvement of autonomic and peripheral nervous systems Degeneration of preganglionic autonomic neurons of the brain stem and spinal cord cause multidomain autonomic failures in MSA [133, 183, 184]. Supraspinal lesions involve cholinergic neurons of the ventrolateral nucleus ambiguous [185, 186], tegmental nuclei [187], ventral periaqueductal dopaminergic neurons [188], medullary and arcuate nucleus, noradrenergic locus ceruleus [134], serotonergic medullary groups, ventrolateral medulla [189], caudal raphe neurons [190, 191], catecholaminergic neurons of rostral ventral medulla, and noradrenergic neurons of the caudal ventrolateral medulla [185, 192]. The medullary serotonergic and catecholaminergic systems are involved in early stages of MSA [193]. Other involved areas are the dorsal vagal nucleus [185], periaqueductal gray [132], the Westphal-Edinger nucleus and posterior hypothalamus, the tuberomamillary and suprachiasmatic nuclei [194], and the pontomedullary reticular formation [149]. The density of αSyn pathology did not correlate with neuronal loss, and there was no correlation between the αSyn burden and disease duration in these regions, indicating that the loss of monoaminergic neurons may progress independently from αSyn accumulation [195]. Sympathetic preganglionic neurons in the intermediolateral cell columns of the thoracolumbar spinal cord [26, 134, 196] and sympathetic ganglia and Schwann cells in autonomic nerves are involved [197]. Neuronal loss affects Onuf's nucleus in the sacral region [198], with minor loss of upper and lower motor neurons [26] and variable involvement of anterior horn cells [134]. Mild degeneration of cardiac sympathetic innervation has been reported in some cases of MSA [199, 200], which accounts for a mild to moderate decrease in the number of tyrosine hydroxylase, but not of neurofilament-immunoreactive nerve fibers in the epicardium. However, depletion of cardiac sympathetic nerves is closely related to the presence of αSyn pathology in the sympathetic ganglia of the CNS [200, 201]. The peripheral nervous system shows αSyn deposits in sympathetic ganglia, skin nerve fibers [138, 202, 203], and Schwann cells [204], but lack of αSyn immunoreactivity in dermal fibers in contrast to PD [203, 205]. Filamentous αSyn aggregates involve the cytoplasm of Schwann cells in cranial, spinal and autonomic nerves in MSA [141, 197, 206]. Clinical features The onset of motor symptoms is 56±9 (mean ± SD) years, with both sexes equally affected [207], however 20-75% of MSA patients have a prodromal/preclinical phase with non-motor symptoms. This phase includes cardiovascular and other autonomic failures (urogenital and sexual dysfunctions, orthostatic hypotension, and REM sleep behavior disorder (RBD), which occurs in 88% or more [208, 209]), which may precede the motor presentation by months to years [210, 211] and indicates more rapid progression of the disease [212, 213]. Average age at disease onset is earlier in MSA-C compared to MSA-P, the latter leading to more severe disability [214-216]. Average duration after clinical diagnosis is 6-10 (mean 9.5) years [12, 23], with few patients surviving more than 15 years [217]. Others have reported a 5 year survival of 78% [218] and a 43% death rate during 3 years of follow-up [135]. A Pan-American multicenter study reported that 68% of the participants presenting as MSA-P showed an age at onset of 61.5 years, and those as MSA-C of 57.4 years [219], while a prospective cohort in the USA reported a median survival of 9.8 (95% CI 8.8-10.7) years [220]. Early autonomic dysfunctions and severity of orthostatic hypertension have negative impact on both disease progression and survival [221] and more than triples the risk of shorter survival [222, 223], and a meta-analysis identified severe dysautonomia, early combined autonomic and motor failure, and early falls as unfavorable predictors of survival, whereas MSA phenotype and sex did not predict survival [224]. Parkinsonism with rigidity, slowness of movements, postural instability, gait disability, and a tendency to fall, characterize the motor presentation of MSA-P [12]. Parkinsonism is rapidly progressing to wheelchair confinement within 5 to 10 years from symptom onset, poorly responsive to L-dopa, and is often associated with atypical features [17]. Unilateral parkinsonism occurs in 40% of MSA patients [220] and typical tremor in 4-10% [225]. Early postural instability and gait difficulties with recurrent falls are also seen in MSA [35]. Polyminimyoclonus, not included in the current diagnostic criteria of MSA, has now been recognized as a specific clinical feature of MSA. Among motor and non-motor symptoms in early MSA, dysarthria was the most prevalent feature (98.4%), followed by sexual dysfunction (95%), RBD (90.2%), constipation (82%), snoring (70.5%), dysphagia (69%), and stridor (42.6%), which was more common in MSA-C than in MSA-P [226]. A resting tremor is rare, whereas irregular postural and action tremor may occur [227, 228]. Cerebellar ataxia, widespread gait, uncoordinated limb movements, action tremor, and spontaneous or gaze invoked nystagmus predominate MSA-C [35]. Hyperreflexia and a Babinski sign occur in 30-50% of patients, while abnormal postures, such as bent spine, antecollis, and hand or foot dystonia are rare [229]. Early generalized and rapidly progressive autonomic failure is typical of MSA [230] and, in the absence of parkinsonism or cerebellar signs, indicating pure autonomic failure, which converts to MSA within a few years in about 28% [231-233]. Among non-motor symptoms observed in 75-95% of patients [234], urinary urgency and increased frequency are common in early disease stages [35]. In a subset of MSA patients with early urinary retention, the disease may begin in the sacral spinal cord and then spread to other regions [235]. Orthostatic hypotension with recurrent syncope, which occurs after the onset of urogenital symptoms, is a hallmark feature of MSA; less specific are dizziness and nausea. Other symptoms are anhydrosis, gastrointestinal dysfunction with early dysphagia and constipation [225], pupillary autonomic involvement with blurred vision and dry eyes. [236]. Dysproportional antecollis and Pisa syndrome are common postural deformities in MSA [35]. About 50% of patients with MSA-P develop cerebellar signs and even a higher proportion of MSA-C cases develop parkinsonian features [23, 220]. Dystonia, repeated falls, drooling, dysphagia, dysphonia, and pain occur in advanced stages of the disease [237]. Laryngeal stridor is rare [210]. Respiratory disturbances including diurnal or nocturnal inspiratory stridor and sleep apnea are frequent [238, 239]. Diagnostic biomarkers Despite numerous studies, to date there are no reliable diagnostic and prognostic biomarkers available. While multimodal imaging of structural and functional brain changes gave insight into the pathophysiology and may evaluate disease progression, recent studies suggest that the combination of neuroimaging and fluid biomarkers may be more successful than using single markers to increase the accuracy of the clinical (differential) diagnosis of MSA [240]. Fluid and tissue biomarkers Studies of αSyn levels in cerebrospinal fluid (CSF) and plasma have been shown to not be useful in the discrimination between MSA and PD or PSP [5, 241, 242]. A recent meta-analysis of available CSF data showed that reduction of p-tau, αSyn, Aβ-42 and total tau and elevated NFL are indicators for MSA [243]. Currently, the most promising approach is a combination of CSF DJ-1, phospho-tau, light chain neurofilament protein (NFL) and Aβ-42 that may be helpful in the differential diagnosis between MSA and other parkinsonian disorders [5, 240, 243, 244] (Fig. 3). Other studies have shown increased CSF levels of cytokines such as MCP-3, MDC, fractalkine, and MIP-1β [246]. Phosphorylated αSyn in red blood cells may be a potential diagnostic biomarker for MSA [247]. The results of proteomics for biomarker discovery and mRNA expression need further elucidation [248].
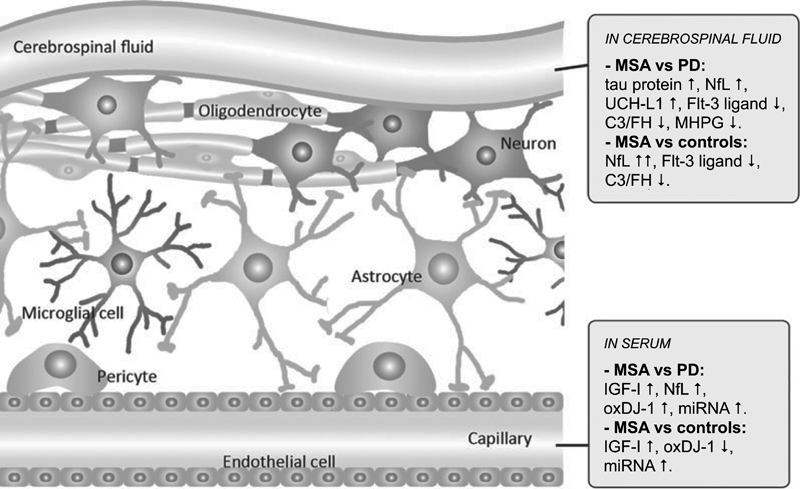
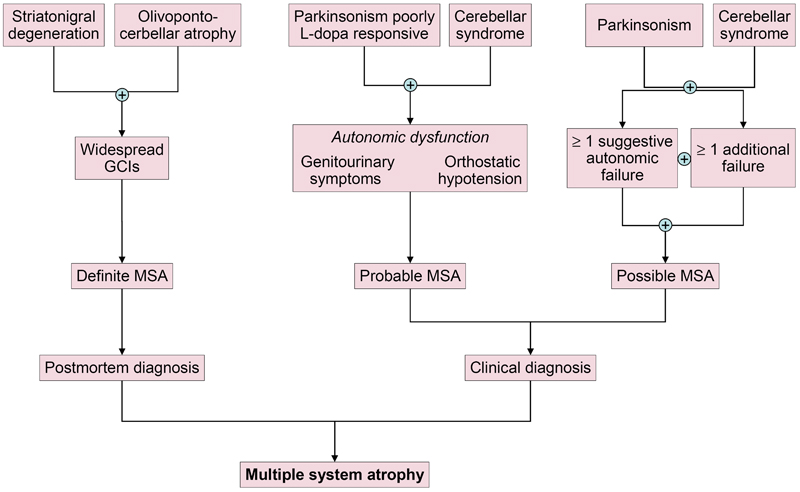
Fig. 3. Candidate biomarkers of multiple system atrophy compared to Parkinson’s disease and controls. Molecular and functional imaging A cardiac sympathetic postganglionic denervation distinguishes PD from MSA, showing intact innervation. I-123 MIBG (metaiodobenzylguanidine) scintigraphy can help differentiate the two diseases with a pooled specificity of 77% (95% CI: 68-84%) [199]. Recent meta-analyses suggest that MIBG imaging is useful to discriminate PD from MSA in moderate to advanced disease stages, but unreliable in early stages [199, 249]. However, interactions with many drugs limit the value of this method [250]. The anteroposterior diameter of the medulla oblongata is a potential imaging marker of parasympathetic dysfunction in MSA [251]. In recent years, several brain magnetic resonance imaging (MRI) features have been described as helpful in the differential diagnosis of parkinsonian syndromes. They include atrophy of the putamen, pons, cerebellum, and middle cerebellar peduncle, a dilated fourth ventricle, and various signal intensity variations on MRI [252]. MRI abnormalities including the "hot-cross bun" sign, a cruciform hyperintensity in the pons [253], and the "putaminal rim sign", which marks hyperintensive bordering of the dorsolateral margins of the puta men in T2-weighted MRI reflecting degeneration and iron deposition, may differentiate MSA-P from PD [254-258]. They are, however, non-specific signs and therefore not included in the recent consensus criteria [3], in contrast to putaminal atrophy which shows 92.3% specificity but low sensitivity (44.4%) [259, 260]. Putaminal atrophy together with hypointense putaminal signal changes on iron-sensitive routine sequences seem to be specific for MSA-P [252]. Others showed significantly increased putaminal diffusivity volumes in the small anterior region of interest in MSA-P versus PD [261]. Another distinguishing feature is the extensive and widespread volume loss across the entire brain in MSA-P [262]. In quantitative MRI studies, the bilateral R2* increase in the putamen best separated MSA-P from PD [263]. Putaminal and infratentorial volume information classified 96.8% of MSA cases [260]. Diffusion tensor imaging permits differentiation between PD and MSA-P, the latter showing higher values of the diffusion coefficient in the inner capsule, corona radiata, and lateral periputaminal white matter [264], while a meta-analysis of putaminal diffusivity measurements showed sensitivity of 90% and specificity of 93% in distinguishing MSA-P from PD based on putaminal diffusivity [265]. Combined use of diffusion ratios and magnetic susceptibility values/quantitative susceptibility mapping allowed differentiation of MSA-P and MSA-C from other parkinsonian syndromes with sensitivities and specificities of 81-100% [266]. Hyperintensity of the middle cerebellar peduncle and hot cross bun sign should be added into the list of additional neuroimaging features of possible MSA-C [182]. Several studies assessed the diagnostic potential of multimodal MRI [267-270]. In conclusion, the sensitivity of conventional MRI findings in MSA compared to PD and healthy controls is inconsistent (36-83%), the specificity of MRI abnormalities differentiating MSA from PD is high (88-100%). Automated imaging differentiation in parkinsonism (AID-P) and magnetic resonance Parkinsonism index (MRPI) are robust biomarkers for PD and MSA [271]. Diffusion weighted images, T2* weighted images and proton density weighted images are useful for diagnosis MSA-P in early stages [272]. Fluorodeoxyglucose-positron emission tomography (FDG-PET) can distinguish MSA-P from PD, showing different patterns of decreased glucose metabolism with a positive predictive value of 97% [273, 274]. Targeting postsynaptic dopaminergic functions using 123FβCIT SPECT differentiates PD (normal or increased signal) from MSA (normal or increased signal) [275]. Dopamine transporter (DAT) imaging showed more prominent and earlier DAT loss in the anterior caudate and ventral putamen in MSA than in PD [276], although normal DAT imaging does not exclude MSA [277]. In autopsy-confirmed cases a greater asymmetry of striatal binding was seen in MSA than in PD [278], but it is highly correlated with SN cell loss [279]. 18F-Dopa-PET showed more widespread BG dysfunction in MSA than in PD without evidence of early compensatory increase in Dopa uptake [280]. Future studies will be needed to determine the usefulness of tau-PET imaging for the characterization of αSyn filaments and the differential diagnosis of atypical parkisonian disorders. Interpretation of tau-PET should be done cautiously, since some MSA cases with severe GCI pathology may be false-positive [281, 282], even though the affinity of PBB3 is 10 to 50 times less than αSyn [283]. 1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinoline carboxamide (PK11195) for imaging microglia-mediated processes showed elevated tracer binding in many areas of the MSA brain, consistent with the known neuropathologic distribution [284]. Diagnostic accuracy and differential diagnosis Revised consensus guidelines define 3 degrees of certainty of clinical diagnosis of MSA: definite, probable and possible [3] (Table 1, Fig. 4).
Fig. 4. Diagnostic scheme for MSA according to the current consensus diagnostic criteria. Definite MSA requires post mortem evidence of widespread αSyn inclusions with concomitant SND or OPCA [1]. Probable MSA is defined as a sporadic, progressive disorder in adults, clinically characterized by severe autonomic failure, urinary dysfunction and poor L-dopa-responsive parkinsonism or cerebellar ataxia. A diagnosis of probable MSA is based on clinical features and ancillary diagnostic tests. Possible MSA can be diagnosed when a sporadic progressive adult-onset disorder with parkinsonism or cerebellar ataxia is accompanied by at least one of the following additional features within 3 years of motor onset: dysphagia, gait ataxia and other cerebellar symptoms (Table 1).
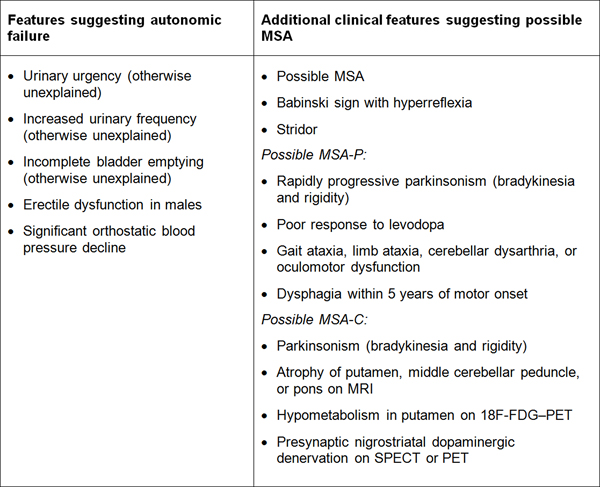
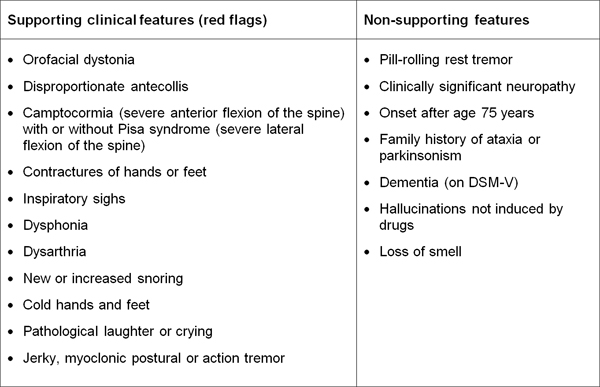
Table 1. Diagnostic clinical markers for MSA. Modified from [240]. "Red flag" diagnostic features The presence of "red flag" (warning sign) features highly specific for MSA may provide important clues for a correct and early diagnosis. They include orofacial dystonia; inspiratory signs, contractures of hands and feet, jerky myoclonic postural/action tremor, polyminimyoclonus, severe dysphonia and dysarthria, pathological laughter and crying, snoring, disproportional antecollis, camptocormia and/or Pisa syndrome, and cold hands and feet [225, 229] (Table 2). In addition, severe disability milestones include: frequent falls, use of urinary catheters, wheelchair dependence, unintelligible speech, cognitive impairment, severe dysphagia, and residential care. In a recent clinicopathological study of 203 clinically diagnosed MSA patients, a lifetime recorded number of red flags in both MSA-P and MSA-C was compared to LBD and PSP [225]. Recognition of patients with early or possible MSA may be supported by one or more red flags, and two or more out of six had a specificity of 98.3% and a sensitivity of 84.2% [228, 229], while no differences were found in the frequencies of red flags within 3 years from disease onset between MSA and MSA look-alikes [225]. Recent studies confirmed the validity of an eight-item pilot scale for the assessment of early MSA [285].
Table 2. Clinical features supporting and non-supporting a diagnosis of multiple system atrophy. Due to the heterogeneity of clinical phenotypes and lack of specific biomarkers, it is a challenge to make a correct antemortem diagnosis of MSA [286]. The sensitivity of the second consensus criteria was 41% for possible and 18% for probable MSA at first clinical visit and 92% and 63% at last clinical visit, respectively [287]. In two recent brain bank studies, among patients diagnosed with MSA during life, only 62% and 79% met the pathological criteria [225, 286], while 25% of patients with the diagnosis of "possible" MSA had different pathological diagnoses, including PD and PSP [225]. The most common misdiagnoses were DLB (13 and 14%, respectively), PSP (6 and 11%) and PD (6%). Autonomic failure was the leading cause of misdiagnosis in PD and DLB, and cerebellar ataxia that of misdiagnosis in PSP [286]. Sporadic spinocerebellar ataxia (SCA) with autonomic failure can masquerade as MSA-C. A study reported that 7% of patients with clinically diagnosed MSA had mutations in SCA genes [288]. Fragile X tremor-ataxia syndrome and X-linked adrenoleukodystrophy can also be misdiagnosed as MSA-C [61]. The possible explanations for the suboptimal diagnostic accuracy of the current consensus criteria for MSA that saw a positive predictive diagnosis even in later disease stages from 60 to 90% [286, 287] have been recently discussed [289]. Atypical MSA Almost all cases of MSA display neuronal loss in both striatonigral and OPC structures [24, 148], with only 11 of 42 cases assigned to the category of "pure" SND [155]. However, MSA has a wider range of presentations, which expands the list of differential diagnoses. Several subtypes of MSA do not fit into the current classification [290]. "Minimal change" MSA is a rare aggressive form with GCIs and neurodegeneration almost restricted to the SN, putamen, and locus coeruleus, thus representing "pure" SND [291-294], suggesting that GCI formation is an early event and may precede neuronal loss. One patient with "minimal" MSA-C showed abundant GCIs in pontine nuclei, middle cerebellar peduncle and cerebellar white matter, with NCIs and neuronal nuclear inclusions restricted to the pontine basis, cerebellar vermis, and inferior olivary nuclei, which were associated with neuronal loss indicating a link between both lesions in early disease [295]. Neurologically normal individuals are rarely found to have GCIs at autopsy as coincidental or incidental findings limited to the pons and inferior olivary nuclei with mild neuronal loss restricted to the SN, suggesting that these regions may be afflicted first in MSA-P [296, 297]. The presence of GCIs may represent an age-related phenomenon not necessarily progressing to overt clinical disease, classifying these cases as "incidental" or "prodromal/preclinical" MSA, similar to incidental LBD [298]. Young-onset MSA with a mean age of 36.4 years shows more L-dopa-induced dyskinesia but less common myoclonus and pyramidal signs compared to late-onset cases. On post mortem analysis, the "minimal change" variant was more common in young-onset MSA [299]. The other extreme are "benign" MSA cases with prolonged survival up to 15 years in about 2-3% of patients [217, 300]. Most of them showed slowly progressing parkinsonism with subsequent rapid deterioration after development of autonomic failure [301]. Many of them developed motor fluctuation and L-dopa-induced choreiform dyskinesias [302, 303]. Other cases of survival up to 18 years revealed extensive distribution of GCIs in the CNS [304]. Another variant of pathological confirmed MSA showed neither parkinsonism nor cerebellar symptoms [305]. An atypical case of frontotemporal lobar degeneration (FTLD)-TDP type A with MSA phenocopy syndrome showed severe striatal degeneration and cerebellar involvement [306], while four cases with clinical features of FTLD, but without autonomic dysfunction, showed frontotemporal atrophy and severe limbic αSyn neuronal pathology with Pick body-like, but tau-negative, inclusions. These cases were suggested to represent a novel subtype of FTLD associated with αSyn (FTLD-αSyn) [307]. Rare cases in a family with pathologic hexanucleotide repeat expansions in C9ORF72, a gene linked to amyotrophic lateral sclerosis, demonstrated clinical and neuroimaging features indistinguishable from MSA [308], and a cerebello-brainstem dominant form of X-linked adrenoleukodystrophy presented as MSA [61]. Recently, rare cases of MSA with transitional or diffuse DLB developing clinical features of PDD or DLB have been reported. Those with neuronal loss in SN but not in striatal or OPC systems with widespread GCIs were considered "minimal change" MSA, in which LBD was considered the primary pathology and MSA as coincidental. APOE allele frequency was not different between these forms [309]. These and other subtypes should be considered in establishing a correct diagnosis of MSA. Cognitive impairment in MSA Unlike other synucleinopathies, MSA has not been associated with significant cognitive impairment (CI), which has been considered an exclusion criterion for the diagnosis of MSA [3]. However, a recent position statement by the Neuropathology Task Force of the Movement Disorder Society indicated that CI may be an under recognized feature in MSA occurring in 17-47% of MSA patients, while severe dementia is rare [310]. Because CI has been underestimated in MSA, not all patients have undergone formal cognitive assessments and, therefore, the frequency could be higher than reported in several studies. The degree of CI in MSA patients ranges from mild to moderate decline and affect executive, attentional and visuospatial functions, while memory is less often impaired [197, 310-312]. CI may occur in early stages of MSA, but it is generally common in advanced cases [313] and often correlates with disease duration [314]. Mild cognitive impairment (MCI) has been reported in up to 40% of MSA-P patients, mainly characterized by frontal dysfunction [310, 315]. Mild or moderate CI has been reported in 14-37% of pathologically proven MSA cases [134, 286, 302, 316]. More severe and widespread cognitive dysfunction was seen in MSA-P than in MSA-C patients [317], probably due to prefrontal impairment [315], whereas others saw no differences in cognitive variables between the two groups [318] or more severe cognitive dysfunctions in MSA-C [319]. CI has been regarded as a result of cortical and subcortical structural changes [320], frontal lobe dysfunction [321, 322], cortical dysfunction driven by focal frontostriatal degeneration [162], alterations in the corpus callosum [323], the dorsolateral prefrontal cortex network [324], or neocortical neuronal loss [159], while others have not found any differences in the severity of pathological findings between cases with and without CI [325]. Recent studies indicated that NCI burden in the hippocampus and parahippocampal gyrus is associated with memory impairment in MSA [326]. Alzheimer’s disease neuropathological changes (ADNC), cerebral amyloid angiopathy (CAA), and cerebrovascular lesions did not differ between cases with and without CI [325], whereas others showed a greater burden of NCIs in medial temporal regions, the hippocampus or perirhinal regions [77, 197, 316, 326, 327]. ADNC has been reported in only 2/35 (7%) autopsy-proven cases of MSA [134], whereas two cases of combined MSA and AD (Braak stages III and VI) have been reported, in which only a few neurons shared αSyn and tau [328]. A recent retrospective clinicopathological study of 48 MSA patients (33 MSA-P and 15 MSA-C) with a mean age at death of 60.5±7.8 (range 46-82) years, reported MCI in 10 cases (20.8%), in which three had associated moderate cortical tau pathology (Braak I-II), and moderate CI in seven patients (14.5%), for which six had associated cortical amyloid plaques and moderate cortical tau pathology (Braak II-III), one had probable primary age-related tauopathy (PART), and one female aged 82 years with severe dementia showed fully developed AD. Cortical Lewy pathology, observed in four cases, was not associated with clinical CI. 77.1% of the MSA cases were free of ADNC, compared to 42% in controls, while Lewy pathology was higher than in the control groups (8.4%) [329]. In view of the limited data on the molecular basis of CI (and other neuropsychiatric symptoms) in MSA, further studies on the pathological basis of CI in MSA are needed. MSA - a prion-like or prion disease? The spread of αSyn pathology from one cell to another and even from one nervous structure to another has been demonstrated in vivo [11, 330-335]. This pattern, resembling prion spreading, has led to the concept of prion-like propagation of αSyn and tau [110]. Self-propagation of αSyn oligomers, however, is not sufficient to declare them as prions, because they show "seeding" activity rather than infectivity of αSyn [336]. However, the applicability of the prion hypothesis in α-synucleinopathies and, in particular, MSA remains controversial, since injections of brain lysated from MSA patients failed to replicate the oligodendroglial αSyn pathology that is typical for MSA. While studies in wild type (wt) mice provided insights into the mechanisms of oligodendroglial αSyn aggregations in MSA, intracerebral inoculation studies in non-human primates to the best of our knowledge have not been performed yet. There are other challenges to the hypothesis that MSA is a prion disease. First, endogenous wt αSyn is insufficient to propagate αSyn pathology; mutant αSyn is needed as a template. The transmission of αSyn "prions" to a second synucleinopathy model and their ability to propagate between two distinct mouse cell lines while retaining strain-specific properties was suggested to provide evidence that MSA is a prion disease [337]. However, these and other mouse experiments have not yet explained why in MSA αSyn pathology predominantly accumulates in oligodendroglia, as MSA-derived αSyn does not appear to have the ability to induce strain-like cell-specific aggregates. This demonstrates that the intrinsic properties of A53T αSyn in the M83 mouse model dominate over any strain features harbored by misfolded αSyn in MSA brains [9]. Furthermore, GCIs have never been identified in wt mouse brains inoculated with MSA-derived αSyn [338]. Hence, αSyn aggregates ("prionoids") derived from MSA patients created a neurodegenerative pattern that is atypical for MSA [336]. Moreover, αSyn aggregates, the morphological hallmarks of MSA, were not detected in MSA-inoculated TgM83+/- mice [339, 340], and no study has definitely propagated patient-derived seeds from cell-to-cell or mouse-to-mouse, or fully characterized αSyn strains from MSA vs. PD [117]. The variety of seeds, animal models, and methodologies currently prevents clear conclusions regarding αSyn-related spreading and toxicity, as well as translation of preclinical findings to human disease [341]. A recent study found no evidence of binding between cellular prion protein (PrPC) and αSyn oligomers, while PrPC neither binds to αSyn oligomers nor mediates their detrimental effects [342]. However, there may be different species of αSyn oligomers, which have different binding capacity with PrPC, and it remains possible that future studies could demonstrate that both PrPC-dependent and -independent pathways could play a role in the pathogenesis of synucleinopathies [343]. Accordingly, it could be possible that aggregated αSyn is potent in cross-seeding of prion protein misfolding and aggregation in vitro, producing self-propagating states that can lead to prion diseases upon serial passing in wt animals [344]. However, recent studies showed that abnormal misfolded cellular prion protein was able to efficiently propagate in the brain of animals even in the absence of αSyn, suggesting that this protein may not act as a key modulator of prion propagation. Thus, αSyn may take part in this process of self-propagation but is not specifically required for sustaining prion conversion and propagation [345]. Finally, gene analyses have shown that the homozygous state of positions 129 in the PRNP gene is not a risk factor for MSA and no variants of the PRNP gene were associated with increased risk for MSA [50]. Review of clinical notes from patients who had died of MSA showed no evidence of neurosurgical transmission [346], and studies of couples whose spouses had autopsy-confirmed PD, PSP, or MSA, did not suggest an increased risk of synucleinopathy development in the other spouses [347, 348]. Although there is no evidence of iatrogenic or direct transmission in autopsy-confirmed MSA cases, this is no evidence of absence of human transmission or misfolded proteins other than prions and β-amyloid, and further research is necessary before any conclusion can be drawn [349]. In conclusion, it seems reasonable to postulate that even if prion-like spreading in experimental systems may justify the view that the progression of neurodegeneration in MSA reflects a cell-to-cell spread of pathological αSyn, this is not sufficient to define MSA as classical prion disease [336]. New therapies So far there are no causative or disease-modifying treatments available for MSA and symptomatic therapies are limited [35, 350]. The first-line treatment of a hypokinetic-rigid syndrome is dopaminergic treatment with L-dopa, the initial responsiveness to which has been reported in 83% of MSA patients [228], but its effect is usually transient, and only 31% showed a response for a period of 3.5 years [23]. L-Dopa response was observed in 42-57% of MSA-P and in 13-25% of MSA-C patients [220]. Recent animal studies suggest that L-dopa failure can be induced by restricted lateral striatal lesions combined with dopaminergic denervation [351]. In some patients, motor fluctuations with wearing-off phenomena or off-bound dystonia were observed [352]. L-dopa-induced dyskinesias were reported in 24.7% of definite MSA patients [23]. Dopamine agonists are not considered a therapeutic option, as they show poor efficacy and may involve severe side effects, particularly the worsening of orthostatic hypotension [353]. For cerebellar symptoms, no efficient drug treatments are available. Deep brain stimulation in MSA patients showed only transient improvement of motor symptoms, but was rapidly counteracted by the occurrence of disabling symptoms [303]. Non-pharmacological treatment options such as physiotherapy and occupational therapy play an important role in improving symptoms and patients' quality of life, and should be integrated into the therapeutic concept [354]. Translational and novel therapeutic approaches Based on the current knowledge about the pathogenesis of MSA and the different findings in animal models, a number of therapeutic strategies have been proposed to target disease progression in MSA [5, 15, 16]. Based on the ability of αSyn to be transferred from cell to cell and to spread through the brain in a prion-like manner, inhibition of αSyn oligomerization and aggregation may constitute a promising therapeutic strategy for disease modification, and interesting efforts have been made in this direction. These include (1) αSyn inhibition, (2) αSyn degradation enhancement, (3) intervening neuroinflammation, and (4) neuronal loss. Numerous randomized, placebo-controlled trials of putative disease-modifying agents have been performed including riluzole, minocyline, lithium, rifampicin, fluoxetine, rasagilin, neuroprotective mesenchymal stem cells, epigallocatechin gallate, intravenous immunoglobulins and others. Although most of these treatments were efficient in cellular or animal models of MSA, in human patients they showed no clinical effects [16, 341]. Among drugs targeting αSyn aggregation, PROMESA studies on the effect of epigallocatechin gallate, a polyphenol found in green tea which reduces aggregation and toxicity of αSyn oligomers [329], did not modify disease progression [355]. Among αSyn degradation enhancing compounds, rapamycin, an autophagy enhancer, showed a reduction of αSyn aggregates in some brain areas [356] in preclinical studies, and is now under clinical trial [357]. Another approach concerns the possible involvement of toll-like receptor 4 (TLR4) and its selective antagonist monophosphoryl lipid A (MPLA) that reduced GCIs and motor deficits in mice [358]. Targeting neuroinflammation, the inhibition of myeloperoxidase as well as the reduction of TNFα-dependent reactions are promising disease-modifying targets and are being clinically tested in MSA patients [16]. The use of microglia inhibitors, such as minocycline, that rescues dopaminergic neurons in MSA mice, and the anti-inflammatory substance fluoxetine, however, fail to change disease progression. An alternative approach was used in MSA patients to target neuroinflammation by delivering intravenous immunoglobulin, but the results were inconclusive [359]. The compound FTY720-Mitoxy, an FDA-approved immunosuppressive for multiple sclerosis, reduced parkinsonism by increasing brain-derived neurotrophic factor (BDNF), and protected movement and mitochondria in wt and CNP-αSyn mice [360]. Numerous efforts have been undertaken to address neuronal loss, including bone marrow-derived mesenchymal stem cells [361, 362] and the antioxidant target of rapamycin (mTOR) receptor [363], however all of these efforts have failed to slow or halt disease progression [16]. Several studies have successfully proven the therapeutic potential of anti-αSyn immunotherapy by preventing αSyn spreading [5, 15]. Based on the fact that active immunization of MBP mice reduced αSyn accumulation and neurodegeneration [364], two αSyn vaccines (PD03A, PD01A) were evaluated in phase I studies with MSA and PD patients and showed good safety and tolerability [365, 366]. Active immunization against αSyn and combination with anti-inflammatory treatment may also be promising therapeutic strategies [367, 368]. Gene therapy may constitute another feasible approach to diminish OS excitotoxicity and subsequent neuronal loss, but none of the used compounds demonstrated effects on disease progression and the underlying neurodegeneration [16]. New strategies targeting αSyn are in progress [16, 280, 369], based on completed or ongoing interventional trials by the MSA coalition [12]. Therefore, there is a strong need to clarify the pathogenic mechanisms of MSA in order to develop new therapeutic strategy options, including combined approaches by targeting different MSA-specific pathogenetic effects. Conclusions and further outlook Current evidence supports the hypothesis that misfolded αSyn contributes to OS that induces a cascade of deleterious events, including proteasomal and mitochondrial dysfunctions, neuroinflammation, and energy failure that is associated with deposition of aberrant αSyn in both glia (mainly oligodendroglia) and neurons resulting in neurodegeneration and demyelination. Currently, the cascade of events that underlies the pathogenesis of MSA is not completely understood. Recent studies using animal models that only partially replicate human pathology and the molecular dynamics of the neurodegenerative process have provided progress in our understanding of MSA pathogenesis. The disease is viewed as a primary synucleinopathy with specific (oligodendro)glial-neuronal degeneration developing secondarily via the oligo-myelin-axon-neuron complex [2, 4, 370]. Strong evidence against a primary neuronal pathology with the formation of GCIs, resulting from secondary accumulation of pathological αSyn that may be of neuronal origin [371], is the fact that GCIs are the hallmark of MSA and not of PD, a disease with similar patterns of αSyn inclusions (LBs) but resulting from different strains of αSyn, differentiating the two disorders [88, 89, 106, 372]. The source of αSyn in MSA and the pathogenic cascade leading to "prion-like" spreading of its strains contributing to progression of the disease need further elucidation, and there is no convincing evidence for the suggestion that MSA is a prion disease. Although disease-modifying treatments are currently not available, better knowledge about the molecular pathogenesis of MSA derived from animal models and human post mortem experience have contributed to the development of future therapeutic strategies to target disease progression in MSA. Acknowledgements The author thanks Mr. E. Mitter-Ferstl, PhD, for secretarial and editorial work. Funding The study was partially funded by the Society for the Promotion of Research in Experimental Neurology, Vienna, Austria. References [1] Trojanowski JQ, Revesz T (2007) Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy, Neuropathol Appl Neurobiol 33:615-20. [2] Jellinger KA, Wenning GK (2016) Multiple system atrophy: pathogenic mechanisms and biomarkers, J Neural Transm (Vienna) 123:555-72. [3] Gilman S et al. (2008) Second consensus statement on the diagnosis of multiple system atrophy, Neurology 71:670-6. [4] Jellinger KA (2018) Multiple system atrophy: an oligodendroglioneural synucleinopathy, J Alzheimers Dis 62:1141-79. [5] Koga S, Dickson DW (2017) Recent advances in neuropathology, biomarkers and therapeutic approach of multiple system atrophy, J Neurol Neurosurg Psychiatry online Aug 31: doi 10.1136/jnnp-2017-315813. [6] Shimohata T (2020) [Diagnosis of multiple system atrophy for establishing disease-modifying therapies] (Japanese), Brain Nerve 72:131-6. [7] Goedert M et al. (2017) The synucleinopathies: Twenty years on, J Parkinsons Dis 7:S53-S71. [8] Monzio Compagnoni G, Di Fonzo A (2019) Understanding the pathogenesis of multiple system atrophy: state of the art and future perspectives, Acta Neuropathol Commun 7:113. [9] Dhillon JS et al. (2019) Dissecting alpha-synuclein inclusion pathology diversity in multiple system atrophy: implications for the prion-like transmission hypothesis, Lab Invest online Feb 8: doi 10.1038/s41374-019-0198-9. [10] Kaji S et al. (2020) Insights into the pathogenesis of multiple system atrophy: focus on glial cytoplasmic inclusions, Transl Neurodegener 9:7. [11] Woerman AL et al. (2018) alpha-Synuclein: multiple system atrophy prions, Cold Spring Harb Perspect Med 8. [12] Krismer F, Wenning GK (2017) Multiple system atrophy: insights into a rare and debilitating movement disorder, Nat Rev Neurol 13:232-43. [13] Refolo V et al. (2018) Progressive striatonigral degeneration in a transgenic mouse model of multiple system atrophy: translational implications for interventional therapies, Acta Neuropathol Commun 6:2. [14] Overk C et al. (2018) Multiple system atrophy: experimental models and reality, Acta Neuropathol 135:33-47. [15] Heras-Garvin A, Stefanova N (2020) MSA: From basic mechanisms to experimental therapeutics, Parkinsonism Relat Disord. [16] Meszaros L et al. (2020) Current symptomatic and disease-modifying treatments in multiple system atrophy, Int J Mol Sci 21. [17] Fanciulli A, Wenning GK (2015) Multiple-system atrophy, N Engl J Med 372:249-63. [18] Linder J et al. (2010) Incidence of Parkinson's disease and parkinsonism in northern Sweden: a population-based study, Mov Disord 25:341-8. [19] Winter Y et al. (2010) Incidence of Parkinson's disease and atypical parkinsonism: Russian population-based study, Mov Disord 25:349-56. [20] Tison F et al. (2000) Prevalence of multiple system atrophy, Lancet 355:495-6. [21] Schrag A et al. (1999) Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study, Lancet 354:1771-5. [22] Chrysostome V et al. (2004) Epidemiology of multiple system atrophy: a prevalence and pilot risk factor study in Aquitaine, France, Neuroepidemiology 23:201-8. [23] Wenning GK et al. (2013) The natural history of multiple system atrophy: a prospective European cohort study, Lancet Neurol 12:264-74. [24] Ozawa T, Onodera O (2017) Multiple system atrophy: clinicopathological characteristics in Japanese patients, Proc Jpn Acad Ser B Phys Biol Sci 93:251-8. [25] Yabe I et al. (2006) MSA-C is the predominant clinical phenotype of MSA in Japan: analysis of 142 patients with probable MSA, J Neurol Sci 249:115-21. [26] Yoshida M (2007) Multiple system atrophy: alpha-synuclein and neuronal degeneration, Neuropathology 27:484-93. [27] Lyoo CH et al. (2008) Effects of disease duration on the clinical features and brain glucose metabolism in patients with mixed type multiple system atrophy, Brain 131:438-46. [28] Itoh K et al. (2014) Definite familial multiple system atrophy with unknown genetics, Neuropathology 34:309-13. [29] Hara K et al. (2007) Multiplex families with multiple system atrophy, Arch Neurol 64:545-51. [30] Hohler AD, Singh VJ (2012) Probable hereditary multiple system atrophy-autonomic (MSA-A) in a family in the United States, J Clin Neurosci 19:479-80. [31] Vidal JS et al. (2010) Familial aggregation in atypical Parkinson's disease: a case control study in multiple system atrophy and progressive supranuclear palsy, J Neurol 257:1388-93. [32] Wüllner U et al. (2004) Probable multiple system atrophy in a German family, J Neurol Neurosurg Psychiatry 75:924-5. [33] Fujioka S et al. (2014) Update on novel familial forms of Parkinson's disease and multiple system atrophy, Parkinsonism Relat Disord 20 Suppl 1:S29-34. [34] Sailer A et al. (2016) A genome-wide association study in multiple system atrophy, Neurology 87:1591-8. [35] Fanciulli A et al. (2019) Multiple system atrophy, Int Rev Neurobiol 149:137-92. [36] Brooks JA et al. (2011) Mutational analysis of parkin and PINK1 in multiple system atrophy, Neurobiol Aging 32:548 e5-7. [37] Gu X et al. (2018) Analysis of GWAS-linked variants in multiple system atrophy, Neurobiol Aging 67:201 e1- e4. [38] Yuan X et al. (2015) An association analysis of the R1628P and G2385R polymorphisms of the LRRK2 gene in multiple system atrophy in a Chinese population, Parkinsonism Relat Disord 21:147-9. [39] Heckman MG et al. (2014) LRRK2 exonic variants and risk of multiple system atrophy, Neurology 83:2256-61. [40] Sklerov M et al. (2017) Frequency of GBA variants in autopsy-proven multiple system atrophy, Mov Disord Clin Pract 4:574-81. [41] Sun QY et al. (2013) Genetic association study of glucocerebrosidase gene L444P mutation in essential tremor and multiple system atrophy in mainland China, J Clin Neurosci 20:217-9. [42] Mitsui J et al. (2015) Variants associated with Gaucher disease in multiple system atrophy, Ann Clin Transl Neurol 2:417-26. [43] Srulijes K et al. (2013) No association of GBA mutations and multiple system atrophy, Eur J Neurol 20:e61-2. [44] Chen X et al. (2016) C9ORF72 repeat expansions in Chinese patients with Parkinson's disease and multiple system atrophy, J Neural Transm (Vienna) 123:1341-5. [45] Al-Chalabi A et al. (2009) Genetic variants of the alpha-synuclein gene SNCA are associated with multiple system atrophy, PLoS One 4:e7114. doi:10.1371/journal.pone.0007114. [46] Scholz SW et al. (2009) SNCA variants are associated with increased risk for multiple system atrophy, Ann Neurol 65:610-4. [47] Federoff M et al. (2016) Genome-wide estimate of the heritability of multiple system atrophy, Parkinsonism Relat Disord 22:35-41. [48] Nussbaum RL (2018) (2017) Genetics of synucleinopathies, Cold Spring Harb Perspect Med 8. [49] Sun Z et al. (2015) SNP rs11931074 of the SNCA gene may not be associated with multiple system atrophy in Chinese population, Int J Neurosci 125:612-5. [50] Chelban V et al. (2017) Analysis of the prion protein gene in multiple system atrophy, Neurobiol Aging 49:216 e15- e18. [51] Ogaki K et al. (2018) Multiple system atrophy and apolipoprotein E, Mov Disord 33:647-50. [52] Laurens B et al. (2017) Multiple system atrophy - State of the art, Curr Neurol Neurosci Rep 17:41. [53] Kiely AP et al. (2013) alpha-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson's disease and multiple system atrophy?, Acta Neuropathol 125:753-69. [54] Kiely AP et al. (2015) Distinct clinical and neuropathological features of G51D SNCA mutation cases compared with SNCA duplication and H50Q mutation, Mol Neurodegener 10:41. [55] Chen Y et al. (2015) Analysis and meta-analysis of five polymorphisms of the LINGO1 and LINGO2 genes in Parkinson's disease and multiple system atrophy in a Chinese population, J Neurol 262:2478-83. [56] Katzeff JS et al. (2019) Cross-examining candidate genes implicated in multiple system atrophy, Acta Neuropathol Commun 7:117. [57] Lin CH et al. (2015) COQ2 gene variants associate with cerebellar subtype of multiple system atrophy in Chinese, Mov Disord 30:436-7. [58] Multiple-System Atrophy Research Collaboration (2013) Mutations in COQ2 in familial and sporadic multiple-system atrophy, N Engl J Med 369:233-44. [59] Zhao Q et al. (2016) Association of the COQ2 V393A variant with risk of multiple system atrophy in East Asians: a case-control study and meta-analysis of the literature, Neurol Sci 37:423-30. [60] Quinzii CM et al. (2014) Mutant COQ2 in multiple-system atrophy, N Engl J Med 371:80-3. doi:10.1056/NEJMc1311763. [61] Ogaki K et al. (2016) Adult-onset cerebello-brainstem dominant form of X-linked adrenoleukodystrophy presenting as multiple system atrophy: case report and literature review, Neuropathology 36:64-76. [62] Sharma M et al. (2014) Mutant COQ2 in multiple-system atrophy, N Engl J Med 371:80-3. [63] Ronchi D et al. (2016) Mutational analysis of COQ2 in patients with MSA in Italy, Neurobiol Aging 45:213 e1-2. [64] Ferguson MC et al. (2014) SHC2 gene copy number in multiple system atrophy (MSA), Clin Auton Res 24:25-30. [65] Barca E et al. (2016) Decreased coenzyme q10 levels in multiple system atrophy cerebellum, J Neuropathol Exp Neurol 75:663-72. [66] Schottlaender LV et al. (2016) Coenzyme Q10 levels are decreased in the cerebellum of multiple-system atrophy patients, PLoS One 11:e0149557. [67] Nakamoto FK et al. (2018) The pathogenesis linked to coenzyme Q10 insufficiency in iPSC-derived neurons from patients with multiple-system atrophy, Sci Rep 8:14215. [68] Mills JD et al. (2015) Transcriptome analysis of grey and white matter cortical tissue in multiple system atrophy, Neurogenetics 16:107-22. [69] Lee ST et al. (2015) Altered expression of miR-202 in cerebellum of multiple-system atrophy, Mol Neurobiol 51:180-6. [70] Wakabayashi K et al. (2016) MicroRNA expression profiles of multiple system atrophy from formalin-fixed paraffin-embedded samples, Neurosci Lett 635:117-22. [71] Mills JD et al. (2016) Strand-specific RNA-sequencing analysis of multiple system atrophy brain transcriptome, Neuroscience 322:234-50. [72] Wan P et al. (2017) The role of long noncoding RNAs in neurodegenerative diseases, Mol Neurobiol 54:2012-21. [73] Sturm E, Stefanova N (2014) Multiple system atrophy: genetic or epigenetic?, Exp Neurobiol 23:277-91. [74] Vidal JS et al. (2008) Risk factors of multiple system atrophy: a case-control study in French patients, Mov Disord 23:797-803. [75] Vanacore N et al. (2005) Case-control study of multiple system atrophy, Mov Disord 20:158-63. [76] Bleasel JM et al. (2016) Animal modeling an oligodendrogliopathy - multiple system atrophy, Acta Neuropathol Commun 4:12. [77] Cykowski MD et al. (2015) Expanding the spectrum of neuronal pathology in multiple system atrophy, Brain 138:2293-309. [78] Valera E, Masliah E (2018) The neuropathology of multiple system atrophy and its therapeutic implications, Auton Neurosci 211:1-6. [79] Puska G et al. (2018) Lysosomal response in relation to alpha-synuclein pathology differs between Parkinson's disease and multiple system atrophy, Neurobiol Dis 114:140-52. [80] Yu Z et al. (2020) Reduced oligodendrocyte exosome secretion in multiple system atrophy involves SNARE dysfunction, Brain. [81] Wenning GK et al. (2008) Multiple system atrophy: a primary oligodendrogliopathy, Ann Neurol 64:239-46. [82] Fellner L et al. (2015) Models of multiple system atrophy, Curr Top Behav Neurosci 22:369-93. [83] Stemberger S et al. (2010) Targeted overexpression of human alpha-synuclein in oligodendroglia induces lesions linked to MSA-like progressive autonomic failure, Exp Neurol 224:459-64. [84] Tanji K et al. (2019) A mouse model of adult-onset multiple system atrophy, Neurobiol Dis 127:339-49. [85] Mavroeidi P et al. (2019) Endogenous oligodendroglial alpha-synuclein and TPPP/p25alpha orchestrate alpha-synuclein pathology in experimental multiple system atrophy models, Acta Neuropathol online Apr 22: doi 10.1007/s00401-019-2014-y. [86] Hayakawa H et al. (2013) Loss of DARPP-32 and calbindin in multiple system atrophy, J Neural Transm 120:1689-98. [86a] Kaji S et al. (2018) Pathological endogenous alpha-synuclein accumulation in oligodendrocyte precursor cells potentially induces inclusions in multiple system atrophy, Stem Cell Reports 10:356-65. [87] Ubhi K et al. (2010) Neurodegeneration in a transgenic mouse model of multiple system atrophy is associated with altered expression of oligodendroglial-derived neurotrophic factors, J Neurosci 30:6236-46. [88] Shahnawaz M et al. (2020) Discriminating alpha-synuclein strains in Parkinson's disease and multiple system atrophy, Nature 578:273-7. [89] Lau A et al. (2020) alpha-Synuclein strains target distinct brain regions and cell types, Nat Neurosci 23:21-31. [90] Morgan SA et al. (2020) alpha-Synuclein filaments from transgenic mouse and human synucleinopathy-containing brains are major seed-competent species, J Biol Chem. [91] Monzio Compagnoni G et al. (2018) Mitochondrial dysregulation and impaired autophagy in iPSC-derived dopaminergic neurons of multiple system atrophy, Stem Cell Reports 11:1185-98. [92] Ubhi K et al. (2014) Widespread microRNA dysregulation in multiple system atrophy - disease-related alteration in miR-96, Eur J Neurosci 39:1026-41. [93] Valera E et al. (2017) MicroRNA-101 modulates autophagy and oligodendroglial alpha-synuclein accumulation in multiple system atrophy, Front Mol Neurosci 10:329. [94] Tanji K et al. (2013) Alteration of autophagosomal proteins in the brain of multiple system atrophy, Neurobiol Dis 49:190-8. [95] Kaindlstorfer C et al. (2018) The relevance of iron in the pathogenesis of multiple system atrophy: a viewpoint, J Alzheimers Dis 61:1253-73. [96] Don AS et al. (2014) Altered lipid levels provide evidence for myelin dysfunction in multiple system atrophy, Acta Neuropathol Commun 2:150. [97] Bleasel JM et al. (2014) Lipid dysfunction and pathogenesis of multiple system atrophy, Acta Neuropathol Commun 2:15. [98] Grigoletto J et al. (2017) Higher levels of myelin phospholipids in brains of neuronal alpha-Synuclein transgenic mice precede myelin loss, Acta Neuropathol Commun 5:37. [99] Stefanova N et al. (2007) Microglial activation mediates neurodegeneration related to oligodendroglial alpha-synucleinopathy: Implications for multiple system atrophy, Mov Disord 22:2196-203. [100] von Bernhardi R et al. (2015) Microglial cell dysregulation in brain aging and neurodegeneration, Front Aging Neurosci 7:124. [101] Miki Y et al. (2017) AMBRA1, a novel alpha-synuclein-binding protein, is implicated in the pathogenesis of multiple system atrophy, Brain Pathol online 2017 March 15: doi 10.1111/bpa.12461. [102] Xiang C et al. (2019) MicroRNAs Dysregulation and Metabolism in Multiple System Atrophy, Front Neurosci 13:1103. [103] Kim T et al. (2019) Alterations in striatal microRNA-mRNA networks contribute to neuroinflammation in multiple system atrophy, Mol Neurobiol 56:7003-21. [104] Ubhi K et al. (2009) Mitochondrial inhibitor 3-nitroproprionic acid enhances oxidative modification of alpha-synuclein in a transgenic mouse model of multiple system atrophy, J Neurosci Res 87:2728-39. [105] Ndayisaba A et al. (2019) TNFalpha inhibitors as targets for protective therapies in MSA: a viewpoint, J Neuroinflammation 16:80. [106] Fearon C, Farrell MA (2020) Disease-specific strains of a-synuclein in multiple system atrophy and Parkinson's disease: but why?, Mov Disord 35:756-7. [107] Guo JL, Lee VM (2014) Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases, Nat Med 20:130-8. [108] Dhillon JS et al. (2019) Comparative analyses of the in vivo induction and transmission of alpha-synuclein pathology in transgenic mice by MSA brain lysate and recombinant alpha-synuclein fibrils, Acta Neuropathol Commun 7:80. [109] Duyckaerts C et al. (2019) The prion-like propagation hypothesis in Alzheimer's and Parkinson's disease, Curr Opin Neurol 32:266-71. [110] Goedert M et al. (2017) Like prions: the propagation of aggregated tau and alpha-synuclein in neurodegeneration, Brain 140:266-78. [111] Karpowicz RJ, Jr. et al. (2019) Transmission of alpha-synuclein seeds in neurodegenerative disease: recent developments, Lab Invest online Feb 13: doi 10.1038/s41374-019-0195-z. [112] Davis AA et al. (2018) Intercellular spread of protein aggregates in neurodegenerative disease, Annu Rev Cell Dev Biol 34:545-68. [113] Valdinocci D et al. (2018) Extracellular Interactions of Alpha-Synuclein in Multiple System Atrophy, Int J Mol Sci 19. [114] Reyes JF et al. (2014) Alpha-synuclein transfers from neurons to oligodendrocytes, Glia 62:387-98. [115] Reyes JF et al. (2019) Binding of alpha-synuclein oligomers to Cx32 facilitates protein uptake and transfer in neurons and oligodendrocytes, Acta Neuropathol 138:23-47. [116] Rossi M et al. (2020) Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies, Acta Neuropathol. [117] Yamasaki TR et al. (2019) Parkinson's disease and multiple system atrophy have distinct alpha-synuclein seed characteristics, J Biol Chem 294:1045-58. [118] Van der Perren A et al. (2020) The structural differences between patient-derived alpha-synuclein strains dictate characteristics of Parkinson's disease, multiple system atrophy and dementia with Lewy bodies, Acta Neuropathol. [119] Bassil F et al. (2017) Viral-mediated oligodendroglial alpha-synuclein expression models multiple system atrophy, Mov Disord 32:1230-9. [120] Heras-Garvin A et al. (2019) Anle138b modulates alpha-synuclein oligomerization and prevents motor decline and neurodegeneration in a mouse model of multiple system atrophy, Mov Disord 34:255-63. [121] Olah J et al. (2017) Role of the microtubule-associated TPPP/p25 in Parkinson's and related diseases and its therapeutic potential, Expert Rev Proteomics 14:301-9. [122] Olah J, Ovadi J (2019) Pharmacological targeting of alpha-synuclein and TPPP/p25 in Parkinson's disease: challenges and opportunities in a Nutshell, FEBS Lett online May 31: doi 10.1002/873-3468.13464. [123] McCormack A et al. (2019) Abundance of synaptic vesicle-related proteins in alpha-synuclein-containing protein inclusions suggests a targeted formation mechanism, Neurotox Res 35:883-97. [124] Rohan Z et al. (2016) Shared and distinct patterns of oligodendroglial response in alpha-synucleinopathies and tauopathies, J Neuropathol Exp Neurol 75:1100-9. [125] Jellinger KA, Lantos PL (2010) Papp-Lantos inclusions and the pathogenesis of multiple system atrophy: an update, Acta Neuropathol 119:657-67. [126] Wenning GK, Jellinger KA (2005) The role of alpha-synuclein in the pathogenesis of multiple system atrophy, Acta Neuropathol 109:129-40. [127] Kübler D et al. (2019) Widespread microglial activation in multiple system atrophy, Mov Disord 34:564-8. [128] Hoffmann A et al. (2019) Oligodendroglial a-synucleinopathy-driven neuroinflammation in multiple system atrophy, Brain Pathol 29:380-96. [129] Kiely AP et al. (2018) Immunohistochemical and molecular investigations show alteration in the inflammatory profile of multiple system atrophy brain, J Neuropathol Exp Neurol 77:598-607. [130] Williams GP et al. (2020) T cell infiltration in both human multiple system atrophy and a novel mouse model of the disease, Acta Neuropathol 139:855-74. [131] VanderHorst VG et al. (2015) alpha-Synuclein pathology accumulates in sacral spinal visceral sensory pathways, Ann Neurol 78:142-9. [132] Benarroch EE et al. (2010) Differential involvement of the periaqueductal gray in multiple system atrophy, Auton Neurosci 158:111-7. [133] Ozawa T (2007) Morphological substrate of autonomic failure and neurohormonal dysfunction in multiple system atrophy: impact on determining phenotype spectrum, Acta Neuropathol (Berl) 114:201-11. [134] Wenning GK et al. (1997) Multiple system atrophy: a review of 203 pathologically proven cases, Mov Disord 12:133-47. [135] Bensimon G et al. (2009) Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: the NNIPPS study, Brain 132:156-71. [136] Wakabayashi K et al. (2010) Involvement of the peripheral nervous system in synucleinopathies, tauopathies and other neurodegenerative proteinopathies of the brain, Acta Neuropathol 120:1-12. [137] Kuzdas-Wood D et al. (2015) Involvement of peripheral nerves in the transgenic plp-alpha-syn model of multiple system atrophy: extending the phenotype, PLoS One 10:e0136575. [138] Donadio V et al. (2020) Skin biopsy may help to distinguishing MSA-P from Parkinson’s disease with orthostatic hypotension, Mov Disord in print. [139] Ahmed Z et al. (2012) The neuropathology, pathophysiology and genetics of multiple system atrophy, Neuropathol Appl Neurobiol 38:4-24. [140] Nakamura K et al. (2015) Accumulation of phosphorylated alpha-synuclein in subpial and periventricular astrocytes in multiple system atrophy of long duration, Neuropathology 36:157-67. [141] Koga S et al. (2017) alpha-synuclein astrogliopathy: A possible specific feature in alpha-synucleinopathy, Neuropathology 37:379-81. [142] Schweighauser M et al. (2020) Structures of alpha-synuclein filaments from multiple system atrophy, Nature, online May 27: doi 10.1038/s41586-020-2317-6. [143] Campbell BC et al. (2001) The solubility of alpha-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinson's disease, J Neurochem 76:87-96. [144] McCormack A et al. (2016) Purification of alpha-synuclein containing inclusions from human post mortem brain tissue, J Neurosci Methods 266:141-50. [145] Sekiya H et al. (2019) Wide distribution of alpha-synuclein oligomers in multiple system atrophy brain detected by proximity ligation, Acta Neuropathol 137:455-66. [146] Kiely AP et al. (2019) Exploring the putative role of kallikrein-6, calpain-1 and cathepsin-D in the proteolytic degradation of alpha-synuclein in multiple system atrophy, Neuropathol Appl Neurobiol 45:347-60. [147] Ozawa T (2006) Pathology and genetics of multiple system atrophy: an approach to determining genetic susceptibility spectrum, Acta Neuropathol 112:531-8. [148] Ozawa T et al. (2004) The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: clinicopathological correlations, Brain 127:2657-71. [149] Papp MI, Lantos PL (1994) The distribution of oligodendroglial inclusions in multiple system atrophy and its relevance to clinical symptomatology, Brain 117:235-43. [150] Inoue M et al. (1997) The distribution and dynamic density of oligodendroglial cytoplasmic inclusions (GCIs) in multiple system atrophy: a correlation between the density of GCIs and the degree of involvement of striatonigral and olivopontocerebellar systems, Acta Neuropathol 93:585-91. [151] Armstrong RA et al. (2004) A quantitative study of the pathological changes in ten patients with multiple system atrophy (MSA), J Neural Transm (Vienna) 111:485-95. [152] Tong J et al. (2015) Low levels of astroglial markers in Parkinson's disease: relationship to alpha-synuclein accumulation, Neurobiol Dis 82:243-53. [153] Ishizawa K et al. (2008) Glial cytoplasmic inclusions and tissue injury in multiple system atrophy: A quantitative study in white matter (olivopontocerebellar system) and gray matter (nigrostriatal system), Neuropathology 28:249-57. [154] Wenning GK et al. (2002) A novel grading scale for striatonigral degeneration (multiple system atrophy), J Neural Transm (Vienna) 109:307-20. [155] Jellinger KA et al. (2005) Grading of neuropathology in multiple system atrophy: proposal for a novel scale, Mov Disord 20 Suppl 12:S29-36. [156] Matsusue E et al. (2008) Putaminal lesion in multiple system atrophy: postmortem MR-pathological correlations, Neuroradiology 50:559-67. [157] Brettschneider J et al. (2018) Converging patterns of alpha-synuclein pathology in multiple system atrophy, J Neuropathol Exp Neurol 77:1005-16. [158] Dickson DW (2012) Parkinson's disease and parkinsonism: neuropathology, Cold Spring Harb Perspect Med 2: a009258. [159] Salvesen L et al. (2015) Changes in total cell numbers of the basal ganglia in patients with multiple system atrophy - A stereological study, Neurobiol Dis 74:104-13. [160] Salvesen L et al. (2017) Neocortical neuronal loss in patients with multiple system atrophy: a stereological study, Cereb Cortex 27:400-10. [161] Brenneis C et al. (2007) Progression of brain atrophy in multiple system atrophy. A longitudinal VBM study, J Neurol 254:191-6. [162] Fiorenzato E et al. (2017) Brain structural profile of multiple system atrophy patients with cognitive impairment, J Neural Transm (Vienna) 124:293-302. [163] Kovacs T et al. (2003) Olfactory bulb in multiple system atrophy, Mov Disord 18:938-42. [164] Koga S, Dickson DW (2018) Recent advances in neuropathology, biomarkers and therapeutic approach of multiple system atrophy, J Neurol Neurosurg Psychiatry 89:175-84. [165] Koga S et al. (2018) Corticobasal degeneration with TDP-43 pathology presenting with progressive supranuclear palsy syndrome: a distinct clinicopathologic subtype, Acta Neuropathol 136:389-404. [166] Koga S et al. (2018) TDP-43 pathology in multiple system atrophy: colocalization of TDP-43 and alpha-synuclein in glial cytoplasmic inclusions, Neuropathol Appl Neurobiol 44:707-21. [167] Robinson JL et al. (2018) Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated, Brain 141:2181-93. [168] Matsusue E et al. (2009) Cerebellar lesions in multiple system atrophy: postmortem MR imaging-pathologic correlations, AJNR Am J Neuroradiol 30:1725-30. [169] Brettschneider J et al. (2017) Progression of alpha-synuclein pathology in multiple system atrophy of the cerebellar type, Neuropathol Appl Neurobiol 43:315-29. [170] Bettencourt C et al. (2020) White matter DNA methylation profiling reveals deregulation of HIP1, LMAN2, MOBP, and other loci in multiple system atrophy, Acta Neuropathol 139:135-56. [171] Nykjaer C et al. (2017) Changes in the cell population in brain white matter in multiple system atrophy, Mov Disord online 2017 Apr 10: doi: 10.1002/mds.26979. [172] Indelicato E et al. (2015) Cerebral autoregulation and white matter lesions in Parkinson's disease and multiple system atrophy, Parkinsonism Relat Disord 21:1393-7. [173] Song YJ et al. (2009) Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression, J Neuropathol Exp Neurol 68:1073-83. [174] Tong J et al. (2010) Brain alpha-synuclein accumulation in multiple system atrophy, Parkinson's disease and progressive supranuclear palsy: a comparative investigation, Brain 133:172-88. [175] Tong J et al. (2017) Brain monoamine oxidase B and A in human parkinsonian dopamine deficiency disorders, Brain 140:2460–74. [176] Gerhard A et al. (2006) In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson's disease, Neurobiol Dis 21:404-12. [177] Ishizawa K et al. (2004) Microglial activation parallels system degeneration in multiple system atrophy, J Neuropathol Exp Neurol 63:43-52. [178] Fellner L, Stefanova N (2013) The role of glia in alpha-synucleinopathies, Mol Neurobiol 47:575-86. [179] Stefanova N et al. (2011) Toll-like receptor 4 promotes alpha-synuclein clearance and survival of nigral dopaminergic neurons, Am J Pathol 179:954-63. [180] Ren S et al. (2019) Altered functional connectivity of cerebello-cortical circuit in multiple system atrophy (cerebellar-type), Front Neurosci 12:996. [181] Shah A et al. (2019) Altered structural connectivity of the motor subnetwork in multiple system atrophy with cerebellar features, Eur Radiol 29:2783-91. [182] Carré G et al. (2020) Brain MRI of multiple system atrophy of cerebellar type: a prospective study with implications for diagnosis criteria, J Neurol 267:1269-77. [183] Coon E et al. (2017) Neuropathology of autonomic dysfunction in synucleinopathies, Mov Disord accepted for publication. [184] Iodice V et al. (2012) Autopsy confirmed multiple system atrophy cases: Mayo experience and role of autonomic function tests, J Neurol Neurosurg Psychiatry 83:453-9. [185] Benarroch EE et al. (2006) Involvement of vagal autonomic nuclei in multiple system atrophy and Lewy body disease, Neurology 66:378-83. [186] Kuzdas D et al. (2013) Oligodendroglial alpha-synucleinopathy and MSA-like cardiovascular autonomic failure: experimental evidence, Exp Neurol 247:531-6. [187] Schmeichel AM et al. (2008) Mesopontine cholinergic neuron involvement in Lewy body dementia and multiple system atrophy, Neurology 70:368-73. [188] Benarroch EE et al. (2008) Loss of A5 noradrenergic neurons in multiple system atrophy, Acta Neuropathol 115:629-34. [189] Benarroch EE et al. (2004) Involvement of medullary serotonergic groups in multiple system atrophy, Ann Neurol 55:418-22. [190] Benarroch EE et al. (2002) Depletion of mesopontine cholinergic and sparing of raphe neurons in multiple system atrophy, Neurology 59:944-6. [191] Benarroch EE et al. (2007) Depletion of putative chemosensitive respiratory neurons in the ventral medullary surface in multiple system atrophy, Brain 130:469-75. [192] Benarroch EE et al. (2015) Putative neuropathological interactions in MSA: focus in the rostral ventrolateral medulla, Clin Auton Res 25:77-80. [193] Tada M et al. (2009) Depletion of medullary serotonergic neurons in patients with multiple system atrophy who succumbed to sudden death, Brain 132:1810-9. [194] Benarroch EE et al. (2015) Histaminergic tuberomammillary neuron loss in multiple system atrophy and dementia with Lewy bodies, Mov Disord 30:1133-9. [195] Coon EA et al. (2016) Medullary neuronal loss is not associated with alpha-synuclein burden in multiple system atrophy, Mov Disord 31:1802-9. [196] Gray F et al. (1988) Quantitative study of lateral horn cells in 15 cases of multiple system atrophy, Acta Neuropathol 75:513-8. [197] Koga S et al. (2017) Profile of cognitive impairment and underlying pathology in multiple system atrophy, Mov Disord 32:405-13. [198] Yamamoto T et al. (2005) When is Onuf's nucleus involved in multiple system atrophy? A sphincter electromyography study, J Neurol Neurosurg Psychiatry 76:1645-8. [199] Treglia G et al. (2011) Diagnostic performance of iodine-123-metaiodobenzylguanidine scintigraphy in differential diagnosis between Parkinson's disease and multiple-system atrophy: a systematic review and a meta-analysis, Clin Neurol Neurosurg 113:823-9. [200] Orimo S et al. (2007) Degeneration of cardiac sympathetic nerve can occur in multiple system atrophy, Acta Neuropathol 113:81-6. [201] Sone M et al. (2005) alpha-Synuclein-immunoreactive structure formation is enhanced in sympathetic ganglia of patients with multiple system atrophy, Acta Neuropathol 110:19-26. [202] Doppler K et al. (2015) Distinctive distribution of phospho-alpha-synuclein in dermal nerves in multiple system atrophy, Mov Disord 30:1688-92. [203] Zange L et al. (2015) Phosphorylated alpha-synuclein in skin nerve fibres differentiates Parkinson's disease from multiple system atrophy, Brain 138:2310-21. [204] Nakamura K et al. (2015) Filamentous aggregations of phosphorylated alpha-synuclein in Schwann cells (Schwann cell cytoplasmic inclusions) in multiple system atrophy, Acta Neuropathol Commun 3:29. [205] Haga R et al. (2015) Clinical utility of skin biopsy in differentiating between Parkinson's disease and multiple system atrophy, Parkinsons Dis 2015:167038. [206] Mori F et al. (2002) Alpha-synuclein immunoreactivity in normal and neoplastic Schwann cells, Acta Neuropathol 103:145-51. [207] Quinn N (1989) Multiple system atrophy--the nature of the beast, J Neurol Neurosurg Psychiatry Suppl:78-89. [208] Iranzo A et al. (2005) Characteristics of idiopathic REM sleep behavior disorder and that associated with MSA and PD, Neurology 65:247-52. [209] Palma JA et al. (2015) Prevalence of REM sleep behavior disorder in multiple system atrophy: a multicenter study and meta-analysis, Clin Auton Res 25:69-75. [210] Jecmenica-Lukic M et al. (2012) Premotor signs and symptoms of multiple system atrophy, Lancet Neurol 11:361-8. [211] Xie T et al. (2015) Comparison of clinical features in pathologically confirmed PSP and MSA patients followed at a tertiary center, npj Parkinson's Dis 1:15007. [212] Giannini G et al. (2020) Progression and prognosis in multiple system atrophy presenting with REM behavior disorder, Neurology 94:e1828-e34. [213] Postuma RB et al. (2009) Quantifying the risk of neurodegenerative disease in idiopathic REM sleep behavior disorder, Neurology 72:1296-300. [214] Coon EA et al. (2015) Clinical features and autonomic testing predict survival in multiple system atrophy, Brain 138:3623-31. [215] Sakushima K et al. (2015) Epidemiology of multiple system atrophy in Hokkaido, the northernmost island of Japan, Cerebellum 14:682-7. [216] Lee SW, Koh SB (2012) Clinical features and disability milestones in multiple system atrophy and progressive supranuclear palsy, J Mov Disord 5:42-7. [217] Petrovic IN et al. (2012) Multiple system atrophy-parkinsonism with slow progression and prolonged survival: a diagnostic catch, Mov Disord 27:1186-90. [218] Jecmenica-Lukic M et al. (2014) Clinical outcomes of two main variants of progressive supranuclear palsy and multiple system atrophy: a prospective natural history study, J Neurol 261:1575-83. [219] Gatto E et al. (2014) Pan-American Consortium of Multiple System Atrophy (PANMSA). A Pan-American multicentre cohort study of Multiple System Atrophy, J Parkinsons Dis 4:693-8. [220] Low PA et al. (2015) Natural history of multiple system atrophy in the USA: a prospective cohort study, Lancet Neurol 14:710-9. [221] Foubert-Samier A et al. (2020) Disease progression and prognostic factors in multiple system atrophy: A prospective cohort study, Neurobiol Dis 139:104813. [222] Figueroa JJ et al. (2014) Multiple system atrophy: prognostic indicators of survival, Mov Disord 29:1151-7. [223] Tada M et al. (2007) Early development of autonomic dysfunction may predict poor prognosis in patients with multiple system atrophy, Arch Neurol 64:256-60. [224] Glasmacher SA et al. (2017) Predictors of survival in progressive supranuclear palsy and multiple system atrophy: a systematic review and meta-analysis, J Neurol Neurosurg Psychiatry 88:402-11. [225] Miki Y et al. (2019) Improving diagnostic accuracy of multiple system atrophy: a clinicopathological study, Brain 142:2813-27. [226] Jung YJ et al. (2020) Various motor and non-motor symptoms in early multiple system atrophy, Neurodegener Dis:1-6. [227] Kaindlstorfer C et al. (2013) Tremor in multiple system atrophy - a review, Tremor Other Hyperkinet Mov (N Y) 3:pii: tre-03-165-4252-1, doi: 10.7916/D8NV9GZ9. [228] Köllensperger M et al. (2010) Presentation, diagnosis, and management of multiple system atrophy in Europe: final analysis of the European multiple system atrophy registry, Mov Disord 25:2604-12. [229] Köllensperger M et al. (2008) Red flags for multiple system atrophy, Mov Disord 23:1093-9. [230] McKay JH, Cheshire WP (2018) First symptoms in multiple system atrophy, Clin Auton Res 28:215-21. [231] Giannini G et al. (2018) The natural history of idiopathic autonomic failure: The IAF-BO cohort study, Neurology 91:e1245-e54. [232] Kaufmann H et al. (2017) Natural history of pure autonomic failure: A United States prospective cohort, Ann Neurol 81:287-97. [233] Singer W et al. (2017) Pure autonomic failure: Predictors of conversion to clinical CNS involvement, Neurology 88:1129-36. [234] Eschlböck S et al. (2017) Non-motor symptoms and gender differences in multiple system atrophy (abstr.), NeuroLogisch Suppl 1:8. [235] Panicker JN et al. (2020) Early presentation of urinary retention in multiple system atrophy: can the disease begin in the sacral spinal cord?, J Neurol 267:659-64. [236] Garcia MD et al. (2018) Ocular features of multiple system atrophy, J Clin Neurosci 47:234-9. [237] Wenning GK et al. (2000) What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson's disease?, J Neurol Neurosurg Psychiatry 68:434-40. [238] Ghorayeb I et al. (2004) Relationship between stridor and sleep apnoea syndrome: is it as simple as that?, J Neurol Neurosurg Psychiatry 75:512-3; author reply 3. [239] Ohshima Y et al. (2017) Natural course and potential prognostic factors for sleep-disordered breathing in multiple system atrophy, Sleep Med 34:13-7. [240] Jellinger KA (2017) Potential clinical utility of multiple system atrophy biomarkers, Expert Rev Neurother 17:1189-208. [241] Aerts MB et al. (2012) CSF alpha-synuclein does not differentiate between parkinsonian disorders, Neurobiol Aging 33:430 e1-3. [242] Mollenhauer B et al. (2011) alpha-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: a cohort study, Lancet Neurol 10:230-40. [243] Cong S et al. (2020) Diagnostic utility of fluid biomarkers in multiple system atrophy: a systematic review and meta-analysis, J Neurol. [244] Laurens B et al. (2015) Fluid biomarkers in multiple system atrophy: A review of the MSA Biomarker Initiative, Neurobiol Dis 80:29-41. [245] Zhang F et al. (2014) Candidate biomarkers of multiple system atrophy in cerebrospinal fluid, Rev Neurosci 25:653-62. [246] Compta Y et al. (2019) Cerebrospinal fluid cytokines in multiple system atrophy: A cross-sectional Catalan MSA registry study, Parkinsonism Relat Disord 65:3-12. [247] Li XY et al. (2020) Phosphorylated Alpha-Synuclein in Red Blood Cells as a Potential Diagnostic Biomarker for Multiple System Atrophy: A Pilot Study, Parkinsons Dis 2020:8740419. [248] Magdalinou NK et al. (2017) Identification of candidate cerebrospinal fluid biomarkers in parkinsonism using quantitative proteomics, Parkinsonism Relat Disord 37:65-71. [249] Orimo S et al. (2012) 123I-MIBG myocardial scintigraphy for differentiating Parkinson's disease from other neurodegenerative parkinsonism: a systematic review and meta-analysis, Parkinsonism Relat Disord 18:494-500. [250] Baschieri F et al. (2017) Iodine-123-meta-iodobenzylguanidine myocardial scintigraphy in isolated autonomic failure: potential red flag for future multiple system atrophy, Front Neurol 8:225. [251] Suzuki M et al. (2020) Relationship between cardiac parasympathetic dysfunction and the anteroposterior diameter of the medulla oblongata in multiple system atrophy, Clin Auton Res. [252] Heim B et al. (2018) Structural imaging in atypical parkinsonism, Int Rev Neurobiol 142:67-148. [253] Schrag A et al. (2000) Differentiation of atypical parkinsonian syndromes with routine MRI, Neurology 54:697-702. [254] Lee JH et al. (2015) Progression of subcortical atrophy and iron deposition in multiple system atrophy: a comparison between clinical subtypes, J Neurol 262:1876-82. [255] Hwang I et al. (2015) Differentiation of parkinsonism-predominant multiple system atrophy from idiopathic Parkinson disease using 3T susceptibility-weighted MR imaging, focusing on putaminal change and lesion asymmetry, AJNR Am J Neuroradiol 36:2227-34. [256] Ramli N et al. (2015) Differentiating multiple-system atrophy from Parkinson's disease, Clin Radiol 70:555-64. [257] Sugiyama A et al. (2015) Putaminal hypointensity on T2*-weighted MR imaging is the most practically useful sign in diagnosing multiple system atrophy: A preliminary study, J Neurol Sci 349:174-8. [258] Deguchi K et al. (2015) Significance of the hot-cross bun sign on T2*-weighted MRI for the diagnosis of multiple system atrophy, J Neurol 262:1433-9. [259] Feng JY et al. (2015) The putaminal abnormalities on 3.0T magnetic resonance imaging: can they separate parkinsonism-predominant multiple system atrophy from Parkinson's disease?, Acta Radiol 56:322-8. [260] Krismer F et al. (2019) Morphometric MRI profiles of multiple system atrophy variants and implications for differential diagnosis, Mov Disord 34:1041-8. [261] De Marzi R et al. (2017) Putaminal diffusion imaging for the differential diagnosis of the parkinsoniao variant of multiple system atrophy from Parkinson's disease: Impact of segmentation accuracy, NeuroLogisch Suppl 1:8-9. [262] Planetta PJ et al. (2015) Distinct functional and macrostructural brain changes in Parkinson's disease and multiple system atrophy, Hum Brain Mapp 36:1165-79. [263] Focke NK et al. (2011) Differentiation of typical and atypical Parkinson syndromes by quantitative MR imaging, AJNR Am J Neuroradiol 32:2087-92. [264] Cnyrim CD et al. (2014) Diffusion tensor imaging in idiopathic Parkinson's disease and multisystem atrophy (Parkinsonian type), Neurodegener Dis 13:1-8. [265] Bajaj S et al. (2017) Diffusion-weighted MRI distinguishes Parkinson disease from the parkinsonian variant of multiple system atrophy: A systematic review and meta-analysis, PLoS One 12:e0189897. [266] Ito K et al. (2017) Differential diagnosis of parkinsonism by a combined use of diffusion kurtosis imaging and quantitative susceptibility mapping, Neuroradiology 59:759-69. [267] Péran P et al. (2018) MRI supervised and unsupervised classification of Parkinson's disease and multiple system atrophy, Mov Disord 33:600-8. [268] Zanigni S et al. (2017) White matter and cortical changes in atypical parkinsonisms: A multimodal quantitative MR study, Parkinsonism Relat Disord 39:44-51. [269] Lee K et al. (2018) LRRK2 p.Ile1371Val mutation in a case with neuropathologically confirmed multi-system atrophy, J Parkinsons Dis 8:93-100. [270] Chelban V et al. (2019) An update on advances in magnetic resonance imaging of multiple system atrophy, J Neurol 266:1036-45. [271] Archer DB et al. (2020) Magnetic resonance imaging and neurofilament light in the differentiation of parkinsonism, Mov Disord. [272] Yamawaki T (2020) [Diagnosis of MSA-P and PSP-P in Early Stage], Brain Nerve 72:331-43. [273] Tang CC, Eidelberg D (2010) Abnormal metabolic brain networks in Parkinson's disease from blackboard to bedside, Prog Brain Res 184:161-76. [274] Grimaldi S et al. (2019) Multiple system atrophy: Phenotypic spectrum approach coupled with brain 18-FDG PET, Parkinsonism Relat Disord 67:3-9. [275] Brooks DJ, Seppi K (2009) Proposed neuroimaging criteria for the diagnosis of multiple system atrophy, Mov Disord 24:949-64. [276] Nocker M et al. (2012) Progression of dopamine transporter decline in patients with the Parkinson variant of multiple system atrophy: a voxel-based analysis of [123I]beta-CIT SPECT, Eur J Nucl Med Mol Imaging 39:1012-20. [277] McKinley J et al. (2014) Normal dopamine transporter imaging does not exclude multiple system atrophy, Parkinsonism Relat Disord 20:933-4. [278] Perju-Dumbrava LD et al. (2012) Dopamine transporter imaging in autopsy-confirmed Parkinson's disease and multiple system atrophy, Mov Disord 27:65-71. [279] Kraemmer J et al. (2014) Correlation of striatal dopamine transporter imaging with post mortem substantia nigra cell counts, Mov Disord 29:1767-73. [280] Levin J et al. (2017) Multiple system atrophy. In: Falup-Pecurariu C, Ferreira J, Martinez-Martin P, Chaudhuri KR (eds) Movement Disorders Curricula. Springer Wien, pp 183-92. [281] Perez-Soriano A et al. (2017) PBB3 imaging in Parkinsonian disorders: Evidence for binding to tau and other proteins, Mov Disord 32:1016-24. [282] Cho H et al. (2017) (18) F-AV-1451 binds to putamen in multiple system atrophy, Mov Disord 32:171-3. [283] Ono M et al. (2017) Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies, Brain 140:764-80. [284] Gerhard A et al. (2003) [11C](R)-PK11195 PET imaging of microglial activation in multiple system atrophy, Neurology 61:686-9. [285] Matsushima M et al. (2017) Validity and reliability of a pilot scale for assessment of multiple system atrophy symptoms, Cerebellum Ataxias 4:11. [286] Koga S et al. (2015) When DLB, PD, and PSP masquerade as MSA: An autopsy study of 134 patients, Neurology 85:404-12. [287] Osaki Y et al. (2009) A validation exercise on the new consensus criteria for multiple system atrophy, Mov Disord 24:2272-6. [288] Kim HJ et al. (2014) Should genetic testing for SCAs be included in the diagnostic workup for MSA?, Neurology 83:1733-8. [289] Stankovic I et al. (2019) A critique of the second consensus criteria for multiple system atrophy, Mov Disord 34:975-84. [290] Watanabe H et al. (2016) Expanding concept of clinical conditions and symptoms in multiple system atrophy, Rinsho Shinkeigaku 56:457-64. [291] Berciano J et al. (2002) Presynaptic parkinsonism in multiple system atrophy mimicking Parkinson's disease: a clinicopathological case study, Mov Disord 17:812-6. [292] Kon T et al. (2013) An autopsy case of preclinical multiple system atrophy (MSA-C), Neuropathology 33:667-72. [293] Wenning GK et al. (1994) Clinical features and natural history of multiple system atrophy. An analysis of 100 cases, Brain 117:835-45. [294] Ling H et al. (2015) Minimal change multiple system atrophy: An aggressive variant?, Mov Disord 30:960-7. [295] Wakabayashi K et al. (2005) An autopsy case of early ("minimal change") olivopontocerebellar atrophy (multiple system atrophy-cerebellar), Acta Neuropathol 110:185-90. [296] Fujishiro H et al. (2008) Glial cytoplasmic inclusions in neurologically normal elderly: prodromal multiple system atrophy?, Acta Neuropathol 116:269-75. [297] Parkkinen L et al. (2007) Abundant glial alpha-synuclein pathology in a case without overt clinical symptoms, Clin Neuropathol 26:276-83. [298] DelleDonne A et al. (2008) Incidental Lewy body disease and preclinical Parkinson disease, Arch Neurol 65:1074-80. [299] Batla A et al. (2018) Young-onset multiple system atrophy: Clinical and pathological features, Mov Disord 33:1099-107. [300] Kim HJ, Jeon BS (2012) Multiple system atrophy with prolonged survival, Mov Disord 27:1834. [301] Calandra-Buonaura G et al. (2013) Multiple system atrophy with prolonged survival: is late onset of dysautonomia the clue?, Neurol Sci 34:1875-8. [302] Kim HJ et al. (2012) Young-onset multiple system atrophy, J Neurol Sci 319:168-70. [303] Meissner WG et al. (2016) Outcome of deep brain stimulation in slowly progressive multiple system atrophy: A clinico-pathological series and review of the literature, Parkinsonism Relat Disord 24:69-75. [304] Masui K et al. (2012) Extensive distribution of glial cytoplasmic inclusions in an autopsied case of multiple system atrophy with a prolonged 18-year clinical course, Neuropathology 32:69-76. [305] Gaig C et al. (2008) Pathological description of a non-motor variant of multiple system atrophy, J Neurol Neurosurg Psychiatry 79:1399-400. [306] Sousa AL et al. (2017) Frontotemporal lobar degeneration-TDP with 'multiple system atrophy phenocopy syndrome', Neuropathol Appl Neurobiol online 2017 Feb 9: doi 10.1111/nan.12391. [307] Aoki N et al. (2015) Atypical multiple system atrophy is a new subtype of frontotemporal lobar degeneration: frontotemporal lobar degeneration associated with alpha-synuclein, Acta Neuropathol 130:93-105. [308] Goldman JS et al. (2014) Multiple system atrophy and amyotrophic lateral sclerosis in a family with hexanucleotide repeat expansions in C9orf72, JAMA Neurol 71:771-4. [309] Koga S et al. (2020) Clinicopathologic and genetic features of multiple system atrophy with Lewy body disease, Brain Pathol. [310] Stankovic I et al. (2014) Cognitive impairment in multiple system atrophy: A position statement by the neuropsychology task force of the MDS multiple system atrophy (MODIMSA) study group, Mov Disord 29:857-67. [311] Abrahao A et al. (2011) Cognitive impairment in multiple system atrophy: Changing concepts, Dement Neuropsychol 5:303-9. [312] Siri C et al. (2013) A cross-sectional multicenter study of cognitive and behavioural features in multiple system atrophy patients of the parkinsonian and cerebellar type, J Neural Transm (Vienna) 120:613-8. [313] Brown RG et al. (2010) Cognitive impairment in patients with multiple system atrophy and progressive supranuclear palsy, Brain 133:2382-93. [314] Hatakeyama M et al. (2018) Predictors of cognitive impairment in multiple system atrophy, J Neurol Sci 388:128-32. [315] Kawai Y et al. (2008) Cognitive impairments in multiple system atrophy: MSA-C vs MSA-P, Neurology 70:1390-6. [316] Homma T et al. (2016) Frequent globular neuronal cytoplasmic inclusions in the medial temporal region as a possible characteristic feature in multiple system atrophy with dementia, Neuropathology 36:421-31. [317] Gatto E et al. (2014) Cognition in a multiple system atrophy series of cases from Argentina, Arq Neuropsiquiatr 72:773-6. [318] Santangelo G et al. (2020) Evolution of neuropsychological profile in motor subtypes of multiple system atrophy, Parkinsonism Relat Disord 70:67-73. [319] Hong HJ et al. (2011) Cognitive impairments in multiple system atrophy of the cerebellar type, J Mov Disord 4:41-5. [320] Caso F et al. (2020) Cognitive impairment and structural brain damage in multiple system atrophy-parkinsonian variant, J Neurol 267:87-94. [321] Cao B et al. (2015) The global cognition, frontal lobe dysfunction and behavior changes in chinese patients with multiple system atrophy, PLoS One 10:e0139773. [322] Chang CC et al. (2009) Cognitive deficits in multiple system atrophy correlate with frontal atrophy and disease duration, Eur J Neurol 16:1144-50. [323] Hara K et al. (2018) Corpus callosal involvement is correlated with cognitive impairment in multiple system atrophy, J Neurol 265:2079-87. [324] Yang H et al. (2020) Altered resting-state voxel-level whole-brain functional connectivity in multiple system atrophy patients with cognitive impairment, Clin Neurophysiol 131:54-62. [325] Asi YT et al. (2014) Neuropathological features of multiple system atrophy with cognitive impairment, Mov Disord 29:884-8. [326] Miki Y et al. (2020) Hippocampal a-synuclein pathology correlates with memory impairment in multiple system atrophy, Brain. [327] Saito M et al. (2017) Perirhinal accumulation of neuronal alpha-synuclein in a multiple system atrophy patient with dementia, Neuropathology 37:431-40. [328] Terni B et al. (2007) Mutant ubiquitin and p62 immunoreactivity in cases of combined multiple system atrophy and Alzheimer's disease, Acta Neuropathol 113:403-16. [329] Jellinger KA (2020) Neuropathological findings in multiple system atrophy with cognitive impairment, J Neural Transm (Vienna). [330] Hasegawa M et al. (2017) Prion-like mechanisms and potential therapeutic targets in neurodegenerative disorders, Pharmacol Ther 172:22-33. [331] Dehay B et al. (2016) Alpha-synuclein propagation: New insights from animal models, Mov Disord 31:161-8. [332] Peelaerts W et al. (2018) a-Synuclein strains and seeding in Parkinson's disease, incidental Lewy body disease, dementia with Lewy bodies and multiple system atrophy: similarities and differences, Cell Tissue Res 373:195-212. [333] Steiner JA et al. (2018) The concept of alpha-synuclein as a prion-like protein: ten years after, Cell Tissue Res 373:161-73. [334] Stopschinski BE, Diamond MI (2017) The prion model for progression and diversity of neurodegenerative diseases, Lancet Neurol 16:323-32. [335] Valdinocci D et al. (2017) Potential modes of intercellular alpha-synuclein transmission, Int J Mol Sci 18:469. [336] Wenning G et al. (2018) Is multiple system atrophy an infectious disease?, Ann Neurol 83:10-2. [337] Woerman AL et al. (2019) Multiple system atrophy prions retain strain specificity after serial propagation in two different Tg(SNCA*A53T) mouse lines, Acta Neuropathol 137:437-54. [338] Watts JC et al. (2013) Transmission of multiple system atrophy prions to transgenic mice, Proc Natl Acad Sci U S A 110:19555-60. [339] Bernis ME et al. (2015) Prion-like propagation of human brain-derived alpha-synuclein in transgenic mice expressing human wild-type alpha-synuclein, Acta Neuropathol Commun 3:75. [340] Prusiner SB et al. (2015) Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism, Proc Natl Acad Sci U S A 112:E5308-17. [341] Meissner WG et al. (2019) Multiple system atrophy: recent developments and future perspectives, Mov Disord. [342] La Vitola P et al. (2019) Cellular prion protein neither binds to alpha-synuclein oligomers nor mediates their detrimental effects, Brain 142:249-54. [343] Gelpi E, Colom-Cadena M (2019) Oligomers: a hot topic for neurodegeneration and a note of caution for experimental models, Brain 142:228-30. [344] Visanji NP et al. (2019) Beyond the synucleinopathies: alpha synuclein as a driving force in neurodegenerative comorbidities, Transl Neurodegener 8:28. [345] Bistaffa E et al. (2019) Prion efficiently replicates in alpha-synuclein knockout mice, Mol Neurobiol 56:7448-57. [346] De Pablo-Fernandez E et al. (2018) No evidence of iatrogenic human transmission in autopsy confirmed multiple system atrophy, Mov Disord 33:1183-4. [347] Coon EA et al. (2019) Conjugal multiple system atrophy: Chance, shared risk factors, or evidence of transmissibility?, Parkinsonism Relat Disord 67:10-3. [348] Rajput AH et al. (2016) Conjugal parkinsonism - Clinical, pathology and genetic study. No evidence of person-to-person transmission, Parkinsonism Relat Disord 31:87-90. [349] Jaunmuktane Z, Brandner S (2019) The role of prion-like mechanisms in neurodegenerative diseases, Neuropathol Appl Neurobiol. [350] Maass S et al. (2016) Current treatment of multiple system atrophy, Curr Treat Options Neurol 18:51. [351] Kaindlstorfer C et al. (2019) L-dopa response pattern in a rat model of mild striatonigral degeneration, PLoS One 14:e0218130. [352] Boesch SM et al. (2002) Dystonia in multiple system atrophy, J Neurol Neurosurg Psychiatry 72:300-3. [353] Flabeau O et al. (2010) Multiple system atrophy: current and future approaches to management, Ther Adv Neurol Disord 3:249-63. [354] Raccagni C et al. (2019) Physiotherapy improves motor function in patients with the Parkinson variant of multiple system atrophy: A prospective trial, Parkinsonism Relat Disord 67:60-5. [355] Levin J et al. (2019) Safety and efficacy of epigallocatechin gallate in multiple system atrophy (PROMESA): a randomised, double-blind, placebo-controlled trial, Lancet Neurol 18:724-35. [356] Lopez-Cuina MG et al. (2018) Rapamycin for treating MSA: a preclinical proof of concept study [abstract], International Congress of Parkinson Disease and Movement Disorders., Mov Disord 33 (suppl 2). [357] Palma J-A et al. (2019) A futility trial of sirolimus in multiple system atrophy: protocol, recruitment and preliminary adverse event profile (abstr.), Neurology 92 (15 Suppl): P3.8-019. [358] Venezia S et al. (2017) Toll-like receptor 4 stimulation with monophosphoryl lipid A ameliorates motor deficits and nigral neurodegeneration triggered by extraneuronal alpha-synucleinopathy, Mol Neurodegener 12:52. [359] Novak P et al. (2012) Treatment of multiple system atrophy using intravenous immunoglobulin, BMC Neurol 12:131. [360] Vidal-Martinez G et al. (2020) FTY720-Mitoxy reduces synucleinopathy and neuroinflammation, restores behavior and mitochondria function, and increases GDNF expression in multiple system atrophy mouse models, Exp Neurol 325:113120. [361] Park KR et al. (2020) Prevention of multiple system atrophy using human bone marrow-derived mesenchymal stem cells by reducing polyamine and cholesterol-induced neural damages, Stem Cell Res Ther 11:63. [362] Singer W et al. (2019) Intrathecal administration of autologous mesenchymal stem cells in multiple system atrophy, Neurology 93:e77-e87. [363] Ndayisaba A, Wenning GK (2020) Inhibition of the mammalian target or rapamycin (mTOR): a potential therapeutic strategy for multiple system atrophy, Clin Auton Res 30:7-8. [364] Mandler M et al. (2015) Active immunization against alpha-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy, Mol Neurodegener 10:10. [365] Lemos M et al. (2020) Targeting a-synuclein by PD03 AFFITOPE® and Anle138b rescues neurodegenerative pathology in a transgenic mouse: clinical relevance, Transl Neurodegener in press. [366] Affiris AG (2018) Affiris announces encouraging long-term data from a series of first-in-human studies using affitope® PD01A targeting oligomeric alpha-synuclein in early Parkinson's disease patients, Affiris Press Release https://affiris.com/investors/#: Press release 14 May 2018. [367] Valera E et al. (2016) Review: Novel treatment strategies targeting alpha-synuclein in multiple system atrophy as a model of synucleinopathy, Neuropathol Appl Neurobiol 42:95-106. [368] Valera E et al. (2017) Combination of alpha-synuclein immunotherapy with anti-inflammatory treatment in a transgenic mouse model of multiple system atrophy, Acta Neuropathol Commun 5:2. [369] Schenk DB et al. (2017) First-in-human assessment of PRX002, an anti-alpha-synuclein monoclonal antibody, in healthy volunteers, Mov Disord 32:211-8. [370] Jellinger KA, Wenning GK (2016) Overlaps between multiple system atrophy and multiple sclerosis: A novel perspective, Mov Disord 31:1767-71. [371] Ubhi K et al. (2011) Multiple system atrophy: a clinical and neuropathological perspective, Trends Neurosci 34:581-90. [372] Halliday GM (2015) Re-evaluating the glio-centric view of multiple system atrophy by highlighting the neuronal involvement, Brain 138:2116-9.
Copyright: © 2020 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |