|
|
|
Free Neuropathology 1:9 (2020) |
|
Original Paper |
|
Neuronal intermediate filament inclusion disease may be incorrectly classified as a subtype of FTLD-FUS |
|
Kevin F. Bieniek1, Keith A. Josephs2, Wen-Lang Lin3, Dennis W. Dickson3 |
|
1 Department of Pathology & Laboratory Medicine, University of Texas Health Science Center, San Antonio, TX, USA |
|
Corresponding author: |
|
Submitted: 02 February 2020 Accepted: 05 March 2020 Copyedited by: Aivi T. Nguyen Published: 11 March 2020 |
|
Keywords: FUS, TDP-43, Atypical frontotemporal lobar degeneration, NIFID, Electron microscope |
|
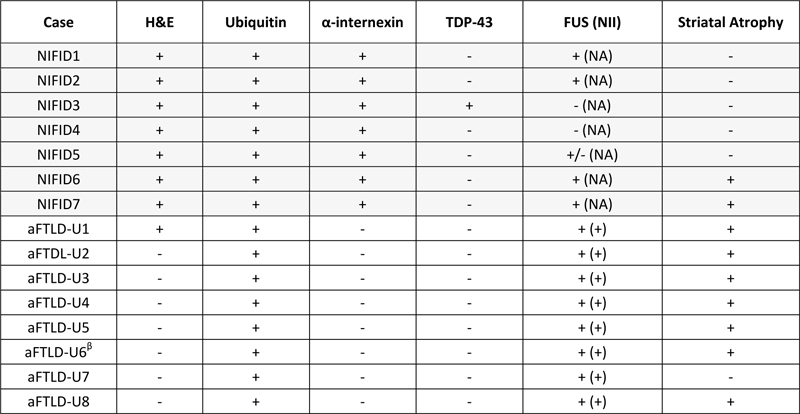
Abstract Background: The majority of cases of frontotemporal lobar degeneration (FTLD) are characterized by focal cortical atrophy with an underlying tau or TDP-43 proteinopathy. A subset of FTLD cases, however, lack tau and TDP-43 immunoreactivity, but have neuronal inclusions positive for ubiquitin, referred to as atypical FTLD (aFTLD-U). Studies have demonstrated that ubiquitin-positive inclusions in aFTLD-U are immunoreactive for fused in sarcoma (FUS). As such, the current nosology for this entity is FTLD-FUS, which is thought to include not only aFTLD-U but also neuronal intermediate filament inclusion disease (NIFID) and basophilic inclusion body disease. Introduction The pathological term frontotemporal lobar degeneration (FTLD) assumes the presence of focal frontal and anterior temporal lobar atrophy(1). Histologically, the majority of FTLDs are pathologically classified into two broad categories of tau-positive FTLD (FTLD-tau) and TDP-43-positive FTLD (FTLD-TDP) based on the presence of tau and TDP-43 immunoreactive inclusions, respectively(2, 3). A subset of FTLD cases however, lack tau and TDP-43 immunoreactivity, and instead have neuronal inclusions that are immunoreactive to ubiquitin(4). Such cases have been referred to in the literature as atypical FTLD with ubiquitin inclusion (aFTLD-U)(5). In the past decade, immunohistochemical studies have revealed that the ubiquitinated protein in cases of aFTLD-U is the fused in sarcoma (FUS) protein(6). Interestingly, there are two other relatively rare FTLD pathological variants that also have neuronal inclusions that are immunoreactive to FUS(7, 8). These include the entity neuronal intermediate filament inclusion disease (NIFID)(9), previously known as neurofilament inclusion body disease(10), and basophilic inclusion body disease(11) that have also been referred to as the generalized variant of Pick’s disease(12). As a result, aFTLD-U, NIFID and basophilic inclusion body disease are all currently classified as subtypes of FTLD-FUS(3, 13). In keeping with pathologically lumping these three entities as FTLD-FUS, is the fact that all three subtypes of FTLD-FUS, particularly aFTLD-U and NIFID, are strongly associated with a clinical presentation of the behavioral variant of frontotemporal dementia (bvFTD)(13-15). Basophilic inclusion body disease is more strongly associated with the juvenile form of amyotrophic lateral sclerosis(16). In addition, aFTLD-U and NIFID have both been found to be associated with striatal atrophy on MRI(17, 18). Hence, aFTLD-U and NIFID have a lot of features in common. One study has directly compared aFTLD-U and NIFID(19). Hence, little is known about which clinical and pathologically differences between the two variants may further help to distinguish them, and whether all cases of aFTLD-U and NIFID do indeed show FUS immunoreactivity. In this study, we set out to address these two unknowns in a cohort of 15 FTLD cases that including aFTLD (n=8) and NIFID (n=7). Materials and methods Subject selection The neuropathological databases at the Mayo Clinic, Jacksonville, Florida were queried to identify all cases of FTLD that had been given a pathological diagnosis of NIFID or aFTLD-U. A total of 15 cases were identified. All 15 cases were evaluated by a single expert neuropathologist (DWD). Clinical data The medical records of all 15 cases were reviewed by one clinician with expertise in neurodegenerative diseases (KAJ) to abstract demographic and clinical information. Data abstracted included sex, age at onset, prominent symptoms during the disease course, family history of any neurodegenerative diseases and final clinical diagnosis prior to death. Pathological methods All 15 cases underwent histologic and ultrastructural evaluation. Tissue sections were stained with hematoxylin and eosin, Luxol fast blue-periodic acid Schiff (LFB-PAS) and Bielschowsky silver stains. Immunohistochemical staining was performed using standard methods. The deparaffinized and rehydrated sections were steamed in distilled water for 30 min and immunostained in batches to assure consistency with a DAKO Autostainer (DAKO, Carpinteria, CA) using 3, 3’diaminobenzidine as the chromogen. After immunostaining, the sections were lightly counterstained with hematoxylin. The following antibodies were used: phosphorylated neurofilament (SMI-31, 1:20,000; Covance, Berkeley, CA); ubiquitin (mouse monoclonal Ubi-1, 1:40,000; EnCor Biotechnology, Alachua, FL; rabbit polyclonal UBQ(20), 1:500 and rabbit polyclonal UH-19(21), 1:2,500); phospho-tau (CP13, 1:100; Peter Davies, Albert Einstein College of Medicine, Bronx, NY); alpha-synuclein (NACP(22), 1:3,000), alpha-internexin (1:100; EnCor Biotechnology, Alachua, FL); TDP-43 (MC2085, Dr. Petrucelli, 1:1500), pTDP-43 (S409/410, Cosmo Bio Co., 1:5000) and rabbit polyclonal anti-FUS (1:500; HPA008784; Sigma, St. Louis. MO and Bethyl Lab; A300-302A; Montgomery, TX). The Sigma antibody gave consistent and better staining and was used throughout the study. The presence or absence of motor neuron disease was assessed and defined as previously described, including stains for activated microglia(23). Electron microscopy Small pieces of formalin-fixed brains were immersed in 2.5% glutaraldehyde-0.1 M cacodylate buffer overnight at 4°C. After washing in buffer, they were post-fixed in aqueous 2% osmium tetroxide for 1 hr, washed and fixed in 1% uranyl acetate-50% ethanol for 30 min, followed by dehydration in 70%, 80%, 95%, 100% ethanols and propylene oxide. They were infiltrated and embedded in Epon 812. Thin sections were stained with uranyl acetate and lead citrate and examined in a Philips 208S electron microscope fitted with a bottom-mount CCD camera (Orius 831, Gatan, Pleasanton, CA). Immunoelectron microscopy Small pieces of formalin-fixed brains were dehydrated in serial washes of 30%, 50%, 70%, 90% ethanol for 10 min each, infiltrated and embedded in LR White resin. They were polymerized in a vacuum oven at 50°C for 2 days. Thin sections were collected on Formvar-coated nickel grids. Grids were floated with section-sides down on citrate buffer, pH 6.0, in a 100°C oven for 10 min, cooled to room temperature for 15 min followed by immunogold labeling. The Sigma anti-FUS was used at 1:20 in PBS. Results Demographics and clinical data for all 15 cases are shown in Table 1. There were 10 males and 5 females with median age at death of 54 years (range 41-69 years). The median disease duration was 5 years (range 3-13 years). The most common final clinical diagnosis in this series was behavioral variant of frontotemporal dementia (bvFTD)(4, 24), rendered in 10 (67%) cases. For the other 5 cases, the final clinical diagnoses were corticobasal syndrome in two cases(25), and one each diagnosed with progressive supranuclear palsy(26), primary lateral sclerosis(27) and multiple system atrophy-Parkinsonian type (MSA-P)(28). Table 1: Demographic and clinical features of all 15 cases
ALS = amyotrophic lateral sclerosis; bvFTD = behavioral variant frontotemporal dementia; CBS = corticobasal syndrome The median age of onset of the NIFID cases was 52 years old (range: 41-61 years) while for aFTLD-U it was 54 years old (range: 42-69 years). Disease duration in NIFID was only 3.5 years (range: 2.0-5.0 years) and was much shorter than the median disease duration of the aFTLD-U group which was 10 years (range: 4-13 years) (P<0.05). The clinical diagnoses were heterogeneous in the NIFID cases, with three cases (43%) diagnosed as bvFTD. On the other hand, of the aFTLD-U cases all but one (88%) had been diagnosed with bvFTD. The NIFID cases were more likely to have had pyramidal tract signs and motor dysfunction compared to the aFTLD-U cases; myoclonic jerks and excessive startle were also observed in NIFID but not aFTLD-U. Pathological findings Gross examination All 15 cases had evidence of frontal and temporal lobe atrophy, and hence all met criteria for FTLD. Striatal atrophy was observed in nine of the 15 cases (Table 2, Figure 1). Table 2: NCI immunohistochemical profile in 15 aFTLD-U and NIFID cases
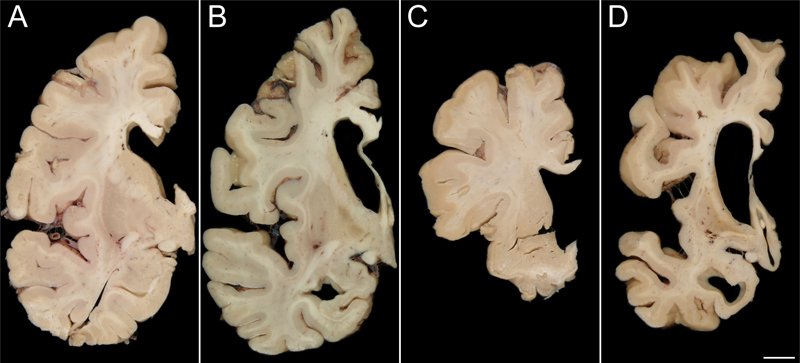
β Diffuse cytoplasmic FUS staining with mini-Pick body-like NCI Figure 1: Striatal atrophy on gross examination
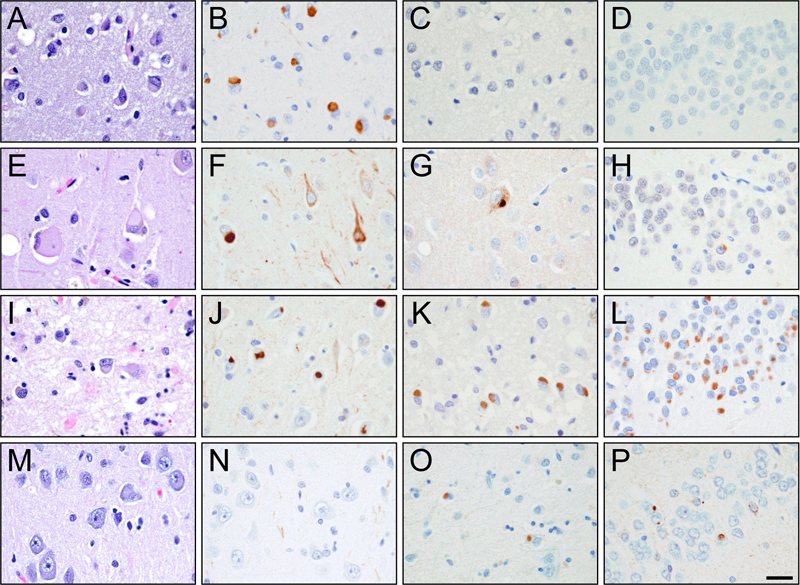
Striatal atrophy was not seen in many NIFID cases (A; Case 5) and was only observed in two cases (B; Case 6). Conversely, only one aFTLD-U case lacked marked striatal atrophy (C; Case 14) while the rest of the aFTLD-U cases demonstrated severe atrophy (D; Case 13) [bar: 1 cm]. Light microscopy Results of the light microscopic examination are shown in Table 2 and Figure 2. None of the 15 cases had evidence of tau deposition, and none met criteria for Alzheimer’s or Lewy body disease. All 15 cases were immune-reactive to ubiquitin. Thirteen of the 15 cases had neuronal inclusions that were immune-reactive to FUS. Seven of the 15 cases had eosinophilic inclusions that were seen on hematoxylin and eosin and were immunostained for α-internexin in keeping with their pathological diagnosis of NIFID. None of the remaining eight cases that had been diagnosed as aFTLD-U showed α-internexin immunoreactivity. Seven of the eight aFTLD-U cases (88%) showed severe striatal atrophy with FUS-positive neuronal cytoplasmic and vermiform intranuclear inclusions. Of the seven NIFID cases, five cases had neuronal inclusions that were immunoreactive to FUS. Unlike in aFTLD-U, striatal atrophy was only present in only 2/7 (29%) of the NIFID cases, both of which were FUS positive. Hence, neither of the two FUS negative NIFID cases had striatal atrophy. Therefore, 9/13 cases with FUS immunoreactive inclusions showed striatal atrophy while 0/2 without FUS showed striatal atrophy. Surprisingly, one of the two NIFID cases without FUS immunoreactivity had lesions that were immunoreactive for both α-internexin and TDP-43 (Figure 3). Figure 2: Alpha-internexin and FUS pathology
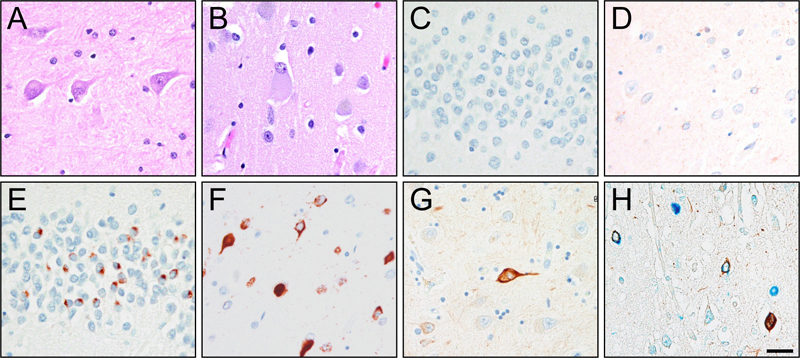
Hematoxylin and Eosin (A/E/I/M), α-internexin (B/F/J/N) and FUS (C/G/K/O) in the frontal cortex and FUS (D/H/L/P) in the hippocampus of Cases 4 (A-D), 5 (E-H), 7 (I-L), and 15 (M-P) [bar:100 μm] Figure 3: FUS negative TDP-43 positive NIFID case
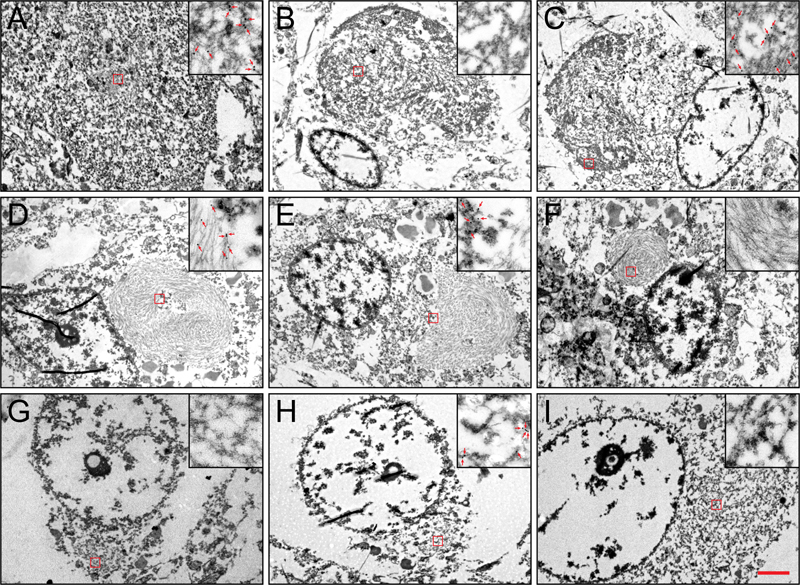
Neuronal eosinophilic (A/B) inclusions in the hippocampus (A/C/E/G) and frontal cortex (B/D/F/H) of Case 3 are negative for FUS (C/D) but positive for TDP-43 (E/F/H), as well as αINX (G/H). α-internexin (brown) and TDP-43 (blue) colocalizes on double-labeling immunohistochemistry (H)[bar:100 μm] Electron and immunoelectron microscopy Results of the electron microscopic examination are shown in Figure 4. In the FUS-positive NIFID cases, we found that FUS was localized to loose granulofilaments that were in close proximity to intermediate filament inclusions that contained tightly packed uncoated filaments unlabeled by FUS antibody. The two types of filaments did not mix. The compact intermediate filament inclusions were similar to those we previously reported(29). In the TDP-positive NIFID case (#3), the TDP-positive NCIs were composed of granulofilaments in tightly packed bundles or loose orientations. Cytoplasmic organelles, e.g. mitochondria, were occasionally encompassed by these inclusions. Figure 4: Electron Microscopy
Variable α-internexin (A/D/G), FUS (B/E/H), and TDP-43 (C/F/I) immunoreactivity on electron microscopy in the neuronal inclusions of Cases 3 (A-C), 2 (D-F), and 9 (G-I)[bar: 2 μm main; 0.3 μm inset] These granulofilaments were not labeled with FUS. The characteristic compact intermediate filament inclusions were not as widespread as the TDP-positive inclusions in this case. Importantly, they were located in separate neurons. In addition to their ultrastructural difference from the TDP-positive inclusions, these intermediate filament inclusions were not labeled by TDP-43. Immunohistochemistry showed that the intermediate filament inclusions were immuno-negative for FUS. The TDP-negative, FUS-negative NIFID case (#4) had compact intermediate filament inclusions similar to those described above. These compact intermediate filament inclusions were immuno-negative to FUS and TDP-43. Neuronal cytoplasmic inclusions in all the aFTLD-U cases consisted of granulofilaments inclusions in loose arrangement and all were immuno-positive for FUS. Discussion In this study we found pathological evidence for aFTLD-U to be a homogeneous entity that is strongly associated with a clinical diagnosis of bvFTD, and pathologically by FUS immunoreactivity and striatal atrophy. On the other hand, we found NIFID to be more heterogeneous with more variable clinical presentations. Furthermore, NIFID does not always appear to be associated with FUS immunoreactivity and is typically not associated with striatal atrophy. Interestingly, we found evidence of overlap between a case that would meet criteria for FTLD-TDP as well as NIFID. In this study, aFTLD-U was a very homogeneous entity and the evidence supports aFTLD-U being classified as FTLD-FUS. From a clinical standpoint aFTLD-U is strongly associated with clinical features of bvFTD as previously reported(14, 30) and hence should be considered in patients presenting with bvFTD especially in the presence of striatal atrophy. Indeed striatal atrophy has been reported in aFTLD-U on antemortem MRI imaging(17). NIFID, on the other hand, as currently defined does not appear to be as distinct an entity clinically and pathologically as aFTLD-U. Supportive of this statement is the fact that of the seven NIFID cases in this study, patients were given five different clinical diagnoses at the last evaluation prior to death. Secondly, striatal atrophy, although present in two NIFID case was absent in the rest. Interestingly, both NIFID cases with striatal atrophy showed FUS immunoreactivity, one with a clinical diagnosis of bvFTD. It therefore appears that striatal atrophy is predictive of FUS, but FUS is not necessarily predictive of striatal atrophy. Regardless, NIFID should also be considered in the differential diagnosis of bvFTD with striatal atrophy and should suggest the presence of FTLD-FUS. The one feature that may be helpful in predicting FTLD-FUS NIFID from FTLD-FUS aFTLD-U in patients with bvFTD and striatal atrophy may be the rapidity of progression, with faster progression being more suggestive of FTLD-FUS NIFID. To understand what may be happening with NIFID it is worth further discussion. Neuronal intermediate filament inclusion body disease is a type of FTLD with previously reported clinical presenting features of bvFTD, corticobasal syndrome, and motor neuron disease, especially the primary lateral sclerosis variant(10, 31, 32). Typically, patients with NIFID have a relatively rapidly progressive course, becoming mute and unable to ambulate, dying around 3 ½ years after onset(10). These features were observed in this cohort of seven NIFID cases. From a pathological standpoint, in NIFID, neuronal cytoplasmic inclusions (NCIs) are easily visible on hematoxylin and eosin, show variable staining to ubiquitin and silver stains but are strikingly immunoreactive to type IV intermediate filaments, including neurofilament and α-internexin(9, 10, 33, 34). These NCI, however, are not morphologically homogeneous and it has been known from the original description of NIFID(10) that NCI appeared to separate into two types: rounded inclusions that are similar to Pick bodies, hence called ‘Pick-body like (PBL) inclusions,’ and smaller more compact inclusions with a glass like appearance called ‘compact hyaline inclusions’(7, 10, 19, 29). Could this NCI inclusion type difference be playing any role in the heterogeneity we observed in this study? The ultrastructual analysis in our seven NIFID cases shows that these two different types of NCI also have different ultrastructual appearances. The PBL inclusions were ultrastructually granulofilamentous while the compact hyaline inclusions had a more tightly compact appearance. With immunoelectron microscopy we found that the granulofilamentous inclusions were immunoreactive to FUS while the compact hyaline inclusions were immunoreactive to intermediate filament. Interestingly all seven NIFID cases had compact hyaline inclusions, and all seven showed α-internexin immunoreactivity. However, unlike in previous reports(35, 36), two of our seven NIFID cases did not show FUS immunoreactivity and one of these two NIFID cases showed predominantly, almost exclusively, compact hyaline inclusions (Case #4). This NIFID case was immuno-negative to FUS, suggesting that the absence of the granulofilamentous inclusions may be the explanation for the absence of FUS immunoreactivity. The other FUS negative case (Case #3) is also unique. In this case, both granulofilamentous inclusions and compact hyaline inclusions were present. As expected, the presence of the compact hyaline inclusions was associated with α-internexin immunoreactivity. Surprisingly though, the granulofilamentous inclusions in this case (Case #3) showed immunoreactivity to TDP-43 but not FUS. It is therefore possible that this is a case of FTLD-TDP in which compact hyaline inclusions happen to also be present and hence accounts for the α-internexin immunoreactivity observed. This would not be counterintuitive since granulofilamentous morphology is the typical appearance of the NCIs in FTLD-TDP(37). On the other hand, we cannot exclude the possibility that this is a case of NIFID with TDP-43 immunoreactivity given that TDP-43 immunoreactivity has been described in many different diseases(38). Compared to our NIFID cases, all eight aFTLD-U cases showed a homogeneous pattern of FUS immunoreactivity similar to what has been previously reported(6, 8, 19, 36). In our eight aFTLD-U cases, ultrastructural analysis demonstrated granulofilamentous inclusions as we previously reported(4), and immunoelectron microscopy revealed FUS immunoreactivity. Of note is the fact that compact hyaline inclusions were absent in all eight aFTLD-U cases. This finding would also support our hypothesis that FUS immunoreactivity in NIFID and aFTLD-U is associated with the presence of the granulofilamentous inclusions while α-internexin immunoreactivity is associated with the presence of the compact hyaline inclusions. It remains unclear however; why some granulofilamentous inclusions show exclusive FUS immunoreactivity (e.g. aFTLD-U) and others show exclusive TDP-43 immunoreactivity (e.g. FTLD-TDP). Only one study to date has reported both FUS and TDP-43 immunoreactivity in the same NCIs(39). Overall, the data from this study support the importance of FUS in the pathogenesis of aFTLD-U and the classification of aFTLD-U as an FTLD-FUS. However, the role of FUS in NIFID is less clear, with evidence supporting NIFID being somewhat different from aFTLD-U with a concern that NIFID, at least not all cases, may be incorrectly classified as a subtype of FTLD-FUS. Supporting this statement is the fact that another FUS negative case with α-internexin positive inclusions has been described(40). In this other case, however, a SOD1 mutation was identified and some inclusions were immunoreactive to SOD1. On a different note, the presence of intermediate filament inclusions and TDP-43 immunoreactive inclusions in the same case makes one contemplate the current sub-classification of FTLD into strict categories. The relationship between FUS and intermediate filament is reminiscent of the relationship between Alzheimer’s disease pathology and Lewy body disease. Alzheimer’s disease pathology can occur in isolation, as can Lewy body disease, but there are instances in which both Alzheimer’s disease and Lewy body disease co-occur. It would be naïve to argue that when two pathologies co-occur, that one pathology is more important or more relevant than the other. In summary, aFTLD-U and NIFID share some but not all features. Striatal atrophy, while appearing to be a characteristic feature of aFTLD-U, does not appear to be a feature of NIFID although it can occur in some cases. Further analyses are needed to better understand the relationship of aFTLD-U to NIFID and of NIFID to FUS. Such analysis should include the assessment of other proteins that have been reported to be associated with aFTLD-U and NIFID including TAF15 and EWS(41), as well as transportin1(42). References 1. Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114(1):5-22. 2. Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 2009;117(1):15-8. 3. Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119(1):1-4. 4. Josephs KA, Lin WL, Ahmed Z, Stroh DA, Graff-Radford NR, Dickson DW. Frontotemporal lobar degeneration with ubiquitin-positive, but TDP-43-negative inclusions. Acta Neuropathol. 2008;116(2):159-67. 5. Mackenzie IR, Foti D, Woulfe J, Hurwitz TA. Atypical frontotemporal lobar degeneration with ubiquitin-positive, TDP-43-negative neuronal inclusions. Brain. 2008;131(Pt 5):1282-93. 6. Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132(Pt 11):2922-31. 7. Neumann M, Roeber S, Kretzschmar HA, Rademakers R, Baker M, Mackenzie IR. Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol. 2009;118(5):605-16. 8. Munoz DG, Neumann M, Kusaka H, Yokota O, Ishihara K, Terada S, et al. FUS pathology in basophilic inclusion body disease. Acta Neuropathol. 2009;118(5):617-27. 9. Cairns NJ, Zhukareva V, Uryu K, Zhang B, Bigio E, Mackenzie IR, et al. alpha-internexin is present in the pathological inclusions of neuronal intermediate filament inclusion disease. Am J Pathol. 2004;164(6):2153-61. 10. Josephs KA, Holton JL, Rossor MN, Braendgaard H, Ozawa T, Fox NC, et al. Neurofilament inclusion body disease: a new proteinopathy? Brain. 2003;126(Pt 10):2291-303. 11. Yokota O, Tsuchiya K, Terada S, Ishizu H, Uchikado H, Ikeda M, et al. Basophilic inclusion body disease and neuronal intermediate filament inclusion disease: a comparative clinicopathological study. Acta Neuropathol. 2008;115(5):561-75. 12. Munoz-Garcia D, Ludwin SK. Classic and generalized variants of Pick's disease: a clinicopathological, ultrastructural, and immunocytochemical comparative study. Ann Neurol. 1984;16(4):467-80. 13. Josephs KA, Hodges JR, Snowden JS, Mackenzie IR, Neumann M, Mann DM, et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. 2011;122(2):137-53. 14. Urwin H, Josephs KA, Rohrer JD, Mackenzie IR, Neumann M, Authier A, et al. FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol. 2010;120(1):33-41. 15. Snowden JS, Hu Q, Rollinson S, Halliwell N, Robinson A, Davidson YS, et al. The most common type of FTLD-FUS (aFTLD-U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS gene. Acta Neuropathol. 2011;122(1):99-110. 16. Matsumoto S, Kusaka H, Murakami N, Hashizume Y, Okazaki H, Hirano A. Basophilic inclusions in sporadic juvenile amyotrophic lateral sclerosis: an immunocytochemical and ultrastructural study. Acta Neuropathol. 1992;83(6):579-83. 17. Josephs KA, Whitwell JL, Parisi JE, Petersen RC, Boeve BF, Jack CR, Jr., et al. Caudate atrophy on MRI is a characteristic feature of FTLD-FUS. Eur J Neurol. 2010;17(7):969-75. 18. Seelaar H, Klijnsma KY, de Koning I, van der Lugt A, Chiu WZ, Azmani A, et al. Frequency of ubiquitin and FUS-positive, TDP-43-negative frontotemporal lobar degeneration. J Neurol. 2010;257(5):747-53. 19. Lashley T, Rohrer JD, Bandopadhyay R, Fry C, Ahmed Z, Isaacs AM, et al. A comparative clinical, pathological, biochemical and genetic study of fused in sarcoma proteinopathies. Brain. 2011;134(Pt 9):2548-64. 20. Dickson DW, Wertkin A, Mattiace LA, Fier E, Kress Y, Davies P, et al. Ubiquitin immunoelectron microscopy of dystrophic neurites in cerebellar senile plaques of Alzheimer's disease. Acta Neuropathol. 1990;79(5):486-93. 21. Lee S, Park YD, Yen SH, Ksiezak-Reding H, Goldman JE, Dickson DW. A study of infantile motor neuron disease with neurofilament and ubiquitin immunocytochemistry. Neuropediatrics. 1989;20(2):107-11. 22. Gwinn-Hardy K, Mehta ND, Farrer M, Maraganore D, Muenter M, Yen SH, et al. Distinctive neuropathology revealed by alpha-synuclein antibodies in hereditary parkinsonism and dementia linked to chromosome 4p. Acta Neuropathol. 2000;99(6):663-72. 23. Josephs KA, Parisi JE, Knopman DS, Boeve BF, Petersen RC, Dickson DW. Clinically undetected motor neuron disease in pathologically proven frontotemporal lobar degeneration with motor neuron disease. Arch Neurol. 2006;63(4):506-12. 24. Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546-54. 25. Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol. 2003;54 Suppl 5:S15-9. 26. Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47(1):1-9. 27. Zhai P, Pagan F, Statland J, Butman JA, Floeter MK. Primary lateral sclerosis: A heterogeneous disorder composed of different subtypes? Neurology. 2003;60(8):1258-65. 28. Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J Neurol Sci. 1989;94(1-3):79-100. 29. Uchikado H, Li A, Lin WL, Dickson DW. Heterogeneous inclusions in neurofilament inclusion disease. Neuropathology. 2006;26(5):417-21. 30. Rohrer JD, Lashley T, Schott JM, Warren JE, Mead S, Isaacs AM, et al. Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain. 2011;134(Pt 9):2565-81. 31. Bigio EH, Lipton AM, White CL, 3rd, Dickson DW, Hirano A. Frontotemporal and motor neurone degeneration with neurofilament inclusion bodies: additional evidence for overlap between FTD and ALS. Neuropathol Appl Neurobiol. 2003;29(3):239-53. 32. Cairns NJ, Grossman M, Arnold SE, Burn DJ, Jaros E, Perry RH, et al. Clinical and neuropathologic variation in neuronal intermediate filament inclusion disease. Neurology. 2004;63(8):1376-84. 33. Josephs KA, Uchikado H, McComb RD, Bashir R, Wszolek Z, Swanson J, et al. Extending the clinicopathological spectrum of neurofilament inclusion disease. Acta Neuropathol. 2005;109(4):427-32. 34. Uchikado H, Shaw G, Wang DS, Dickson DW. Screening for neurofilament inclusion disease using alpha-internexin immunohistochemistry. Neurology. 2005;64(9):1658-9. 35. Armstrong RA, Gearing M, Bigio EH, Cruz-Sanchez FF, Duyckaerts C, Mackenzie IR, et al. The spectrum and severity of FUS-immunoreactive inclusions in the frontal and temporal lobes of ten cases of neuronal intermediate filament inclusion disease. Acta Neuropathol. 2011;121(2):219-28. 36. Mackenzie IR, Munoz DG, Kusaka H, Yokota O, Ishihara K, Roeber S, et al. Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol. 2011;121(2):207-18. 37. Lin WL, Dickson DW. Ultrastructural localization of TDP-43 in filamentous neuronal inclusions in various neurodegenerative diseases. Acta Neuropathol. 2008;116(2):205-13. 38. Josephs KA, Mackenzie I, Frosch MP, Bigio EH, Neumann M, Arai T, et al. LATE to the PART-y. Brain. 2019;142(9):e47. 39. Deng HX, Zhai H, Bigio EH, Yan J, Fecto F, Ajroud K, et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann Neurol. 2010;67(6):739-48. 40. Nakamura M, Bieniek KF, Lin WL, Graff-Radford NR, Murray ME, Castanedes-Casey M, et al. A truncating SOD1 mutation, p.Gly141X, is associated with clinical and pathologic heterogeneity, including frontotemporal lobar degeneration. Acta Neuropathol. 2015;130(1):145-57. 41. Neumann M, Bentmann E, Dormann D, Jawaid A, DeJesus-Hernandez M, Ansorge O, et al. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain. 2011;134(Pt 9):2595-609. 42. Brelstaff J, Lashley T, Holton JL, Lees AJ, Rossor MN, Bandopadhyay R, et al. Transportin1: a marker of FTLD-FUS. Acta Neuropathol. 2011;122(5):591-600.
Copyright: © 2020 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |